Lu Peirong, Li Longbiao, Wu Yu, Mukaida Naofumi, Zhang Xueguang
Clinical Immunology Key Laboratory of Jiangsu Province, the First Affilated Hospital of Soochow University, Suzhou, China.
Mol Vis. 2008 Sep 5;14:1614-22.
To evaluate the roles of CCL3 and its specific chemokine receptors, CCR1 and CCR5, in alkali-induced corneal neovascularization (CNV).
Chemical denudation of corneal and limbal epithelium was performed on wild-type (WT) BALB/c mice and CCL3-, CCR1-, and CCR5-deficienct (knockout [KO]) counterparts. Two weeks after injury CNV was quantified by immunostaining with anti-CD31. Angiogenic factor expression and leukocyte accumulation in the early phase after injury were quantified by reverse transcription polymerase chain reaction (RT-PCR) and immunohistochemical analysis, respectively.
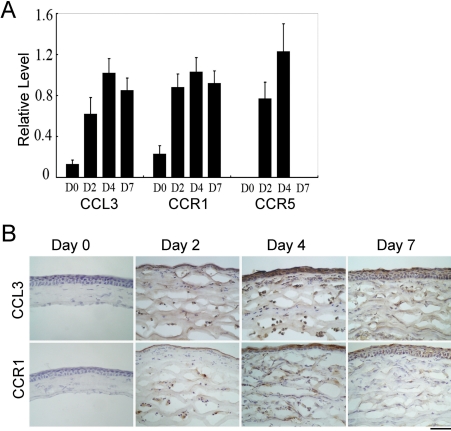
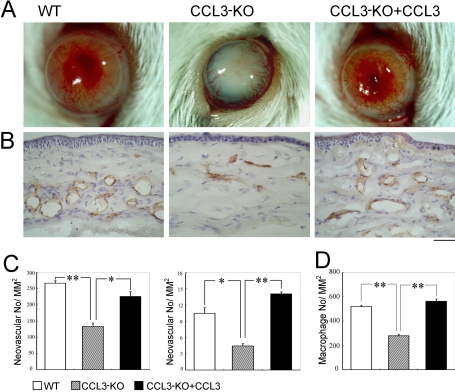
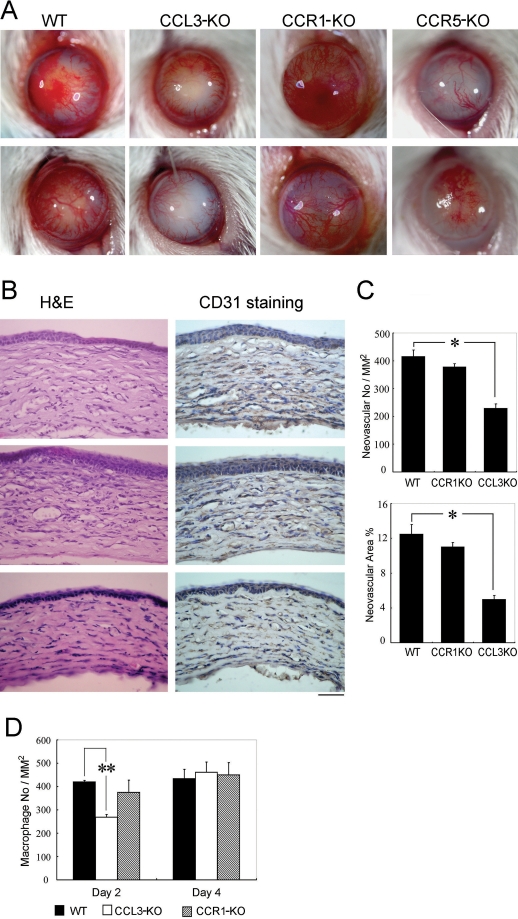
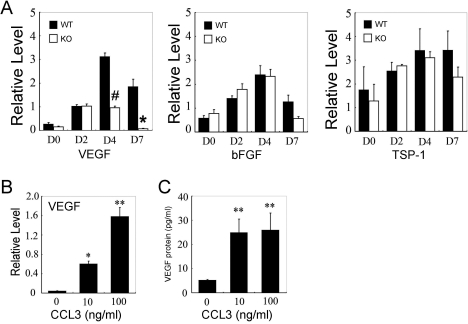
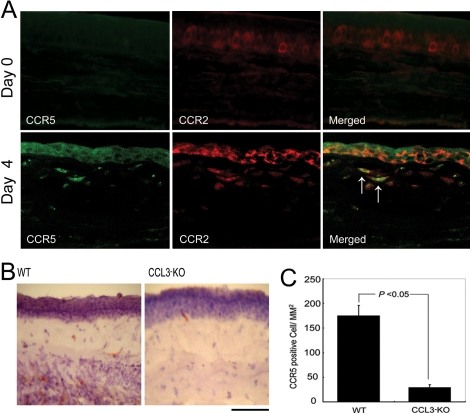
Alkali injury augmented the intraocular mRNA expression of CCL3 and its receptors, CCR1 and CCR5, together with a transient infiltration of F4/80 positive macrophages and Gr-1 positive neutrophils. Compared with WT mice, CCL3-KO and CCR5-KO mice but not CCR1-KO mice exhibited reduced CNV two weeks after injury both macroscopically and microscopically as evidenced by CD31 positive areas. Concomitantly, the infiltration of F4/80 positive macrophages but not Gr-1 positive neutrophils was significantly attenuated in CCL3-KO mice compared with WT mice. Intracorneal infiltration of CCR5 expressing cells was significantly impaired in CCL3-KO mice compared with WT mice. Alkali injury induced a massive increase in the intraocular mRNA expression of a potent angiogenic factor, vascular endothelial growth factor (VEGF), in WT mice whereas these increments were severely retarded in CCL3-KO mice. Moreover, CCL3 enhanced VEGF expression by murine peritoneal macrophages at both the mRNA and the protein level. Furthermore, topical CCL3 application restored CNV, which was macroscopically and microscopically reduced in CCL3-KO mice after two weeks to levels similar to those found in WT mice.
In alkali-induced CNV, CCL3 induced macrophages to infiltrate and produce VEGF by binding to CCR5 but not to CCR1 and eventually promoted angiogenesis.
评估CCL3及其特异性趋化因子受体CCR1和CCR5在碱诱导的角膜新生血管化(CNV)中的作用。
对野生型(WT)BALB/c小鼠以及CCL3、CCR1和CCR5缺陷型(基因敲除[KO])小鼠进行角膜和角膜缘上皮的化学剥脱。损伤两周后,用抗CD31免疫染色对CNV进行定量。分别通过逆转录聚合酶链反应(RT-PCR)和免疫组织化学分析对损伤早期血管生成因子的表达和白细胞聚集进行定量。
碱损伤增加了CCL3及其受体CCR1和CCR5的眼内mRNA表达,同时伴有F4/80阳性巨噬细胞和Gr-1阳性中性粒细胞的短暂浸润。与WT小鼠相比,CCL3-KO和CCR5-KO小鼠(而非CCR1-KO小鼠)在损伤两周后,宏观和微观上均表现出CNV减少,CD31阳性区域可证明这一点。同时,与WT小鼠相比,CCL3-KO小鼠中F4/80阳性巨噬细胞的浸润显著减弱,但Gr-1阳性中性粒细胞的浸润未减弱。与WT小鼠相比,CCL3-KO小鼠中表达CCR5的细胞的角膜内浸润明显受损。碱损伤使WT小鼠眼内一种强效血管生成因子血管内皮生长因子(VEGF)的mRNA表达大量增加,而在CCL3-KO小鼠中这些增加则严重受阻。此外,CCL3在mRNA和蛋白质水平上均增强了小鼠腹腔巨噬细胞的VEGF表达。此外,局部应用CCL3可恢复CNV,两周后CCL3-KO小鼠宏观和微观上减少的CNV恢复到与WT小鼠相似的水平。
在碱诱导的CNV中,CCL3通过与CCR5而非CCR1结合,诱导巨噬细胞浸润并产生VEGF,最终促进血管生成。