Livny Jonathan, Teonadi Hidayat, Livny Miron, Waldor Matthew K
Channing Laboratories, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, United States of America.
PLoS One. 2008 Sep 12;3(9):e3197. doi: 10.1371/journal.pone.0003197.
Diverse bacterial genomes encode numerous small non-coding RNAs (sRNAs) that regulate myriad biological processes. While bioinformatic algorithms have proven effective in identifying sRNA-encoding loci, the lack of tools and infrastructure with which to execute these computationally demanding algorithms has limited their utilization. Genome-wide predictions of sRNA-encoding genes have been conducted in less than 3% of all sequenced bacterial strains, leading to critical gaps in current annotations. The relative paucity of genome-wide sRNA prediction represents a critical gap in current annotations of bacterial genomes and has limited examination of larger issues in sRNA biology, such as sRNA evolution.
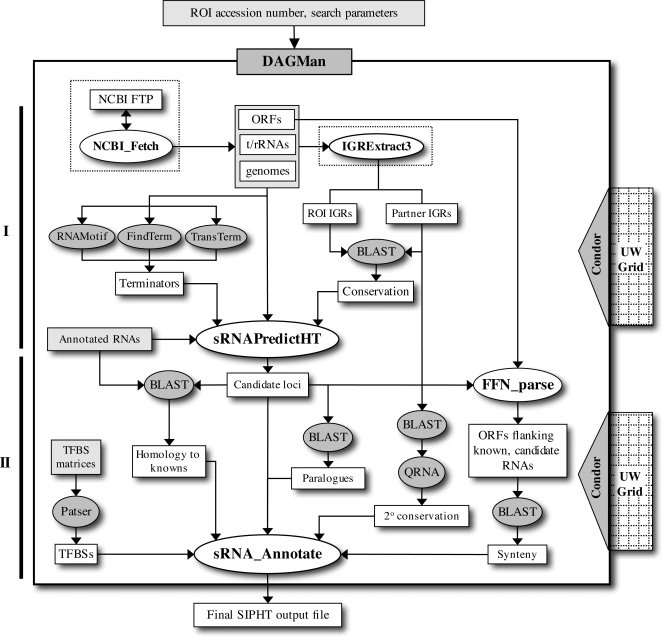
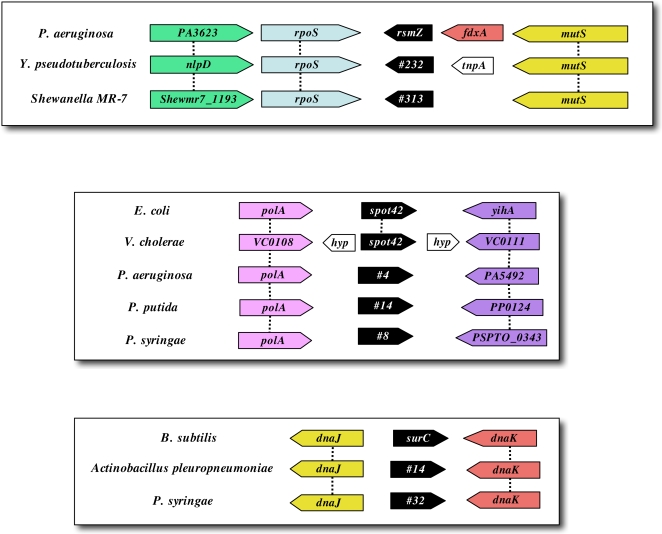
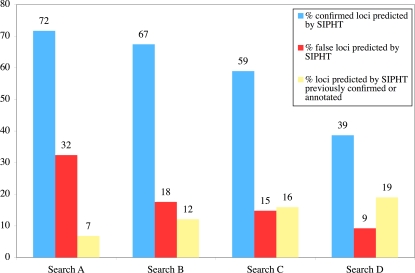
METHODOLOGY/PRINCIPAL FINDINGS: We have developed and deployed SIPHT, a high throughput computational tool that utilizes workflow management and distributed computing to effectively conduct kingdom-wide predictions and annotations of intergenic sRNA-encoding genes. Candidate sRNA-encoding loci are identified based on the presence of putative Rho-independent terminators downstream of conserved intergenic sequences, and each locus is annotated for several features, including conservation in other species, association with one of several transcription factor binding sites and homology to any of over 300 previously identified sRNAs and cis-regulatory RNA elements. Using SIPHT, we conducted searches for putative sRNA-encoding genes in all 932 bacterial replicons in the NCBI database. These searches yielded nearly 60% of previously confirmed sRNAs, hundreds of previously annotated cis-encoded regulatory RNA elements such as riboswitches, and over 45,000 novel candidate intergenic loci.
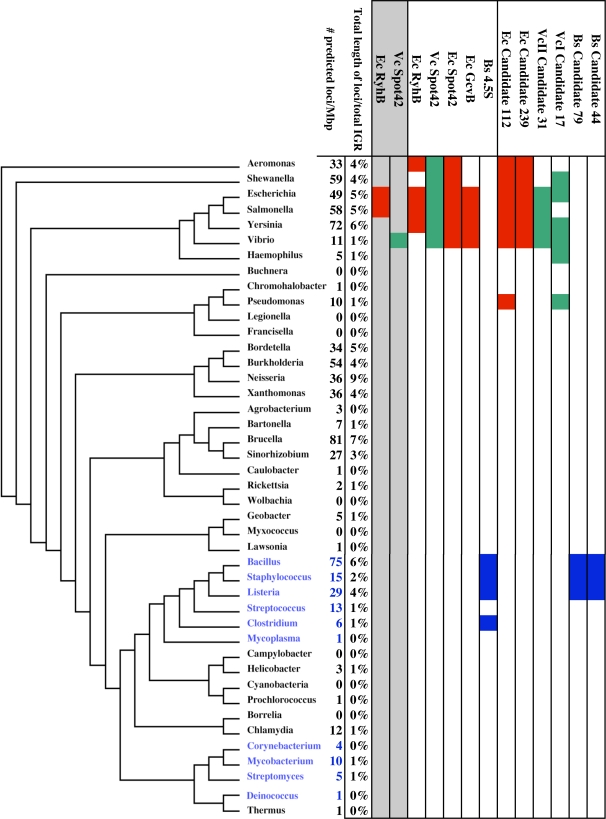
CONCLUSIONS/SIGNIFICANCE: Candidate loci were identified across all branches of the bacterial evolutionary tree, suggesting a central and ubiquitous role for RNA-mediated regulation among bacterial species. Annotation of candidate loci by SIPHT provides clues into the potential biological function of thousands of previously confirmed and candidate regulatory RNAs and affords new insights into the evolution of bacterial riboregulation.
多种细菌基因组编码大量调控众多生物学过程的小非编码RNA(sRNA)。虽然生物信息学算法已被证明在识别编码sRNA的基因座方面有效,但缺乏执行这些计算要求高的算法的工具和基础设施限制了它们的应用。在所有已测序的细菌菌株中,对编码sRNA基因进行全基因组预测的比例不到3%,导致当前注释存在关键空白。全基因组sRNA预测相对较少,这是当前细菌基因组注释中的一个关键空白,限制了对sRNA生物学中更大问题的研究,如sRNA进化。
方法/主要发现:我们开发并部署了SIPHT,这是一种高通量计算工具,利用工作流管理和分布式计算有效地进行全基因组范围内基因间编码sRNA基因的预测和注释。基于保守基因间序列下游假定的不依赖Rho的终止子的存在来识别候选编码sRNA的基因座,并且每个基因座针对几个特征进行注释,包括在其他物种中的保守性、与几种转录因子结合位点之一的关联以及与300多个先前鉴定的sRNA和顺式调控RNA元件中的任何一个的同源性。使用SIPHT,我们在NCBI数据库中的所有932个细菌复制子中搜索假定的编码sRNA的基因。这些搜索产生了近60%先前已确认的sRNA、数百个先前注释的顺式编码调控RNA元件(如核糖开关)以及超过45000个新的候选基因间基因座。
结论/意义:在细菌进化树的所有分支中都鉴定出了候选基因座,这表明RNA介导的调控在细菌物种中起着核心且普遍的作用。SIPHT对候选基因座的注释为数千个先前已确认和候选的调控RNA的潜在生物学功能提供了线索,并为细菌核糖调控的进化提供了新的见解。