Gaffney Daniel J, Keightley Peter D
McGill University and Genome Québec Innovation Centre, 740 ave Dr Penfield Rm 7208, Montréal, Québec, H3A 1A4, Canada.
BMC Evol Biol. 2008 Sep 30;8:265. doi: 10.1186/1471-2148-8-265.
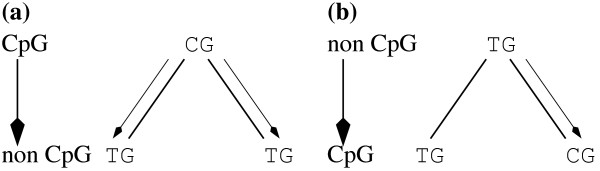
Molecular evolutionary studies in mammals often estimate nucleotide substitution rates within and outside CpG dinucleotides separately. Frequently, in alignments of two sequences, the division of sites into CpG and non-CpG classes is based simply on the presence or absence of a CpG dinucleotide in either sequence, a procedure that we refer to as CpG/non-CpG assignment. Although it likely that this procedure is biased, it is generally assumed that the bias is negligible if species are very closely related.
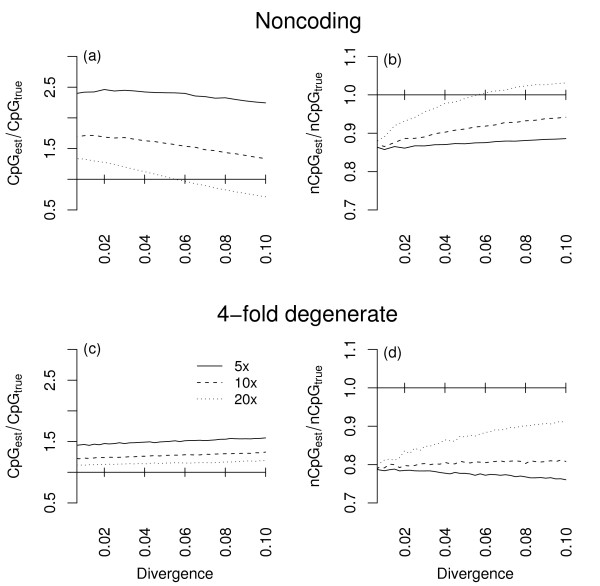
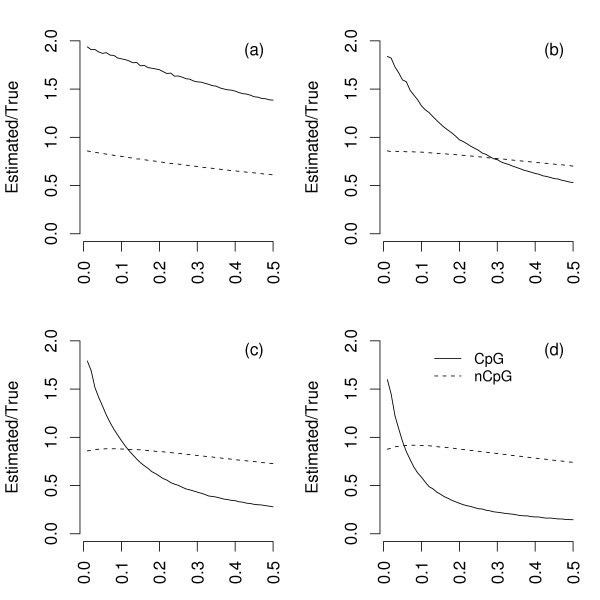
Using simulations of DNA sequence evolution we show that assignment of the ancestral CpG state based on the simple presence/absence of the CpG dinucleotide can seriously bias estimates of the substitution rate, because many true non-CpG changes are misassigned as CpG. Paradoxically, this bias is most severe between closely related species, because a minimum of two substitutions are required to misassign a true ancestral CpG site as non-CpG whereas only a single substitution is required to misassign a true ancestral non-CpG site as CpG in a two branch tree. We also show that CpG misassignment bias differentially affects fourfold degenerate and noncoding sites due to differences in base composition such that fourfold degenerate sites can appear to be evolving more slowly than noncoding sites. We demonstrate that the effects predicted by our simulations occur in a real evolutionary setting by comparing substitution rates estimated from human-chimp coding and intronic sequence using CpG/non-CpG assignment with estimates derived from a method that is largely free from bias.
Our study demonstrates that a common method of assigning sites into CpG and non CpG classes in pairwise alignments is seriously biased and recommends against the adoption of ad hoc methods of ancestral state assignment.
哺乳动物的分子进化研究通常分别估计CpG二核苷酸内部和外部的核苷酸替代率。在两条序列的比对中,位点划分为CpG和非CpG类别通常仅仅基于两条序列中是否存在CpG二核苷酸,我们将此过程称为CpG/非CpG分类。尽管此过程可能存在偏差,但一般认为,如果物种亲缘关系非常近,这种偏差可以忽略不计。
通过DNA序列进化模拟,我们发现基于CpG二核苷酸的简单存在与否来确定祖先CpG状态会严重影响替代率的估计,因为许多真正的非CpG变化被错误地归类为CpG。矛盾的是,这种偏差在亲缘关系近的物种之间最为严重,因为在两棵分支的树中,将一个真正的祖先CpG位点错误归类为非CpG至少需要两次替代,而将一个真正的祖先非CpG位点错误归类为CpG只需要一次替代。我们还表明,由于碱基组成的差异,CpG错误分类偏差对四倍简并位点和非编码位点的影响不同,使得四倍简并位点看起来比非编码位点进化得更慢。通过比较使用CpG/非CpG分类从人类-黑猩猩编码和内含子序列估计的替代率与从基本无偏差的方法得出的估计值,我们证明了模拟预测的效应在实际进化环境中确实存在。
我们的研究表明,在成对比对中将位点划分为CpG和非CpG类别的常用方法存在严重偏差,并建议不要采用临时的祖先状态分类方法。