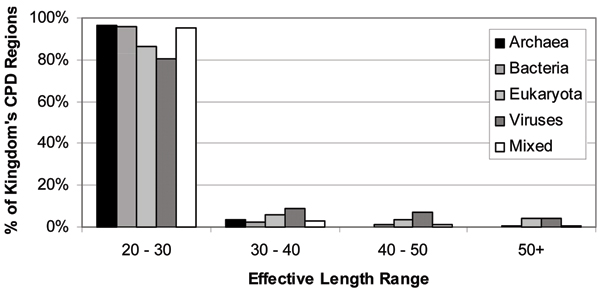
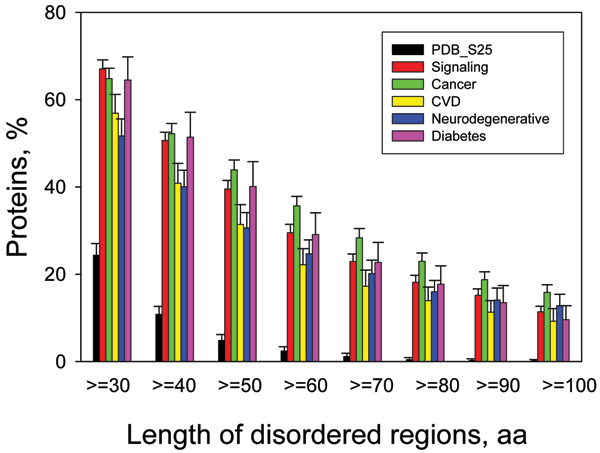
Dunker A Keith, Oldfield Christopher J, Meng Jingwei, Romero Pedro, Yang Jack Y, Chen Jessica Walton, Vacic Vladimir, Obradovic Zoran, Uversky Vladimir N
Center for Computational Biology and Bioinformatics, Indiana University Schools of Medicine and Informatics, Indianapolis, IN 46202, USA.
BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S1. doi: 10.1186/1471-2164-9-S2-S1.
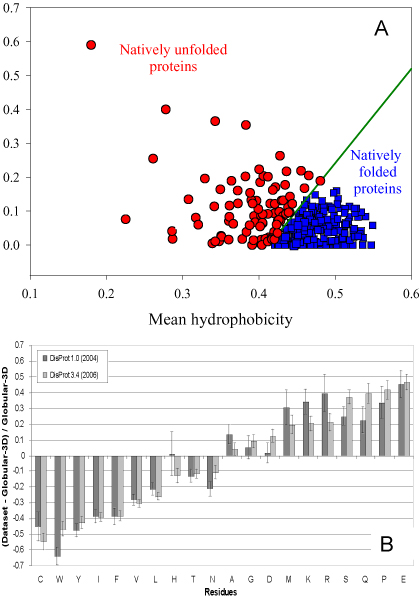
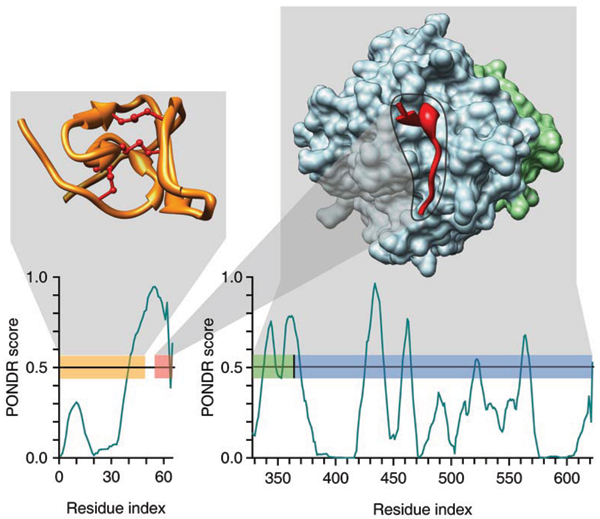
Our first predictor of protein disorder was published just over a decade ago in the Proceedings of the IEEE International Conference on Neural Networks (Romero P, Obradovic Z, Kissinger C, Villafranca JE, Dunker AK (1997) Identifying disordered regions in proteins from amino acid sequence. Proceedings of the IEEE International Conference on Neural Networks, 1: 90-95). By now more than twenty other laboratory groups have joined the efforts to improve the prediction of protein disorder. While the various prediction methodologies used for protein intrinsic disorder resemble those methodologies used for secondary structure prediction, the two types of structures are entirely different. For example, the two structural classes have very different dynamic properties, with the irregular secondary structure class being much less mobile than the disorder class. The prediction of secondary structure has been useful. On the other hand, the prediction of intrinsic disorder has been revolutionary, leading to major modifications of the more than 100 year-old views relating protein structure and function. Experimentalists have been providing evidence over many decades that some proteins lack fixed structure or are disordered (or unfolded) under physiological conditions. In addition, experimentalists are also showing that, for many proteins, their functions depend on the unstructured rather than structured state; such results are in marked contrast to the greater than hundred year old views such as the lock and key hypothesis. Despite extensive data on many important examples, including disease-associated proteins, the importance of disorder for protein function has been largely ignored. Indeed, to our knowledge, current biochemistry books don't present even one acknowledged example of a disorder-dependent function, even though some reports of disorder-dependent functions are more than 50 years old. The results from genome-wide predictions of intrinsic disorder and the results from other bioinformatics studies of intrinsic disorder are demanding attention for these proteins.
Disorder prediction has been important for showing that the relatively few experimentally characterized examples are members of a very large collection of related disordered proteins that are wide-spread over all three domains of life. Many significant biological functions are now known to depend directly on, or are importantly associated with, the unfolded or partially folded state. Here our goal is to review the key discoveries and to weave these discoveries together to support novel approaches for understanding sequence-function relationships.
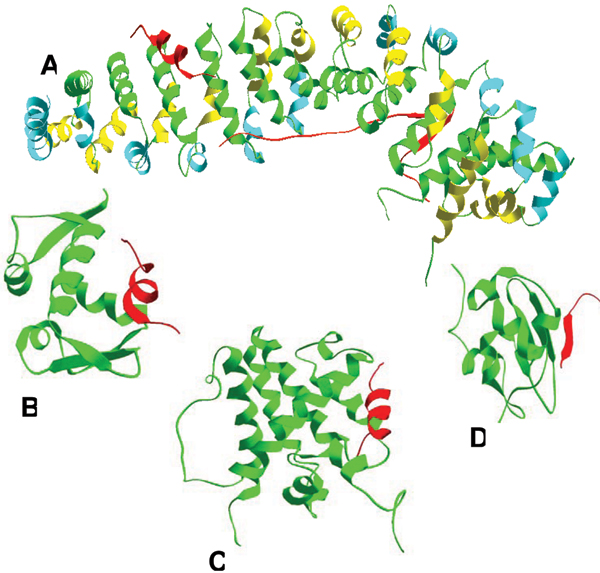
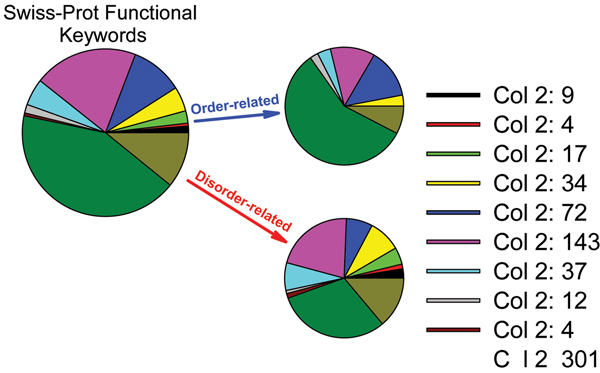
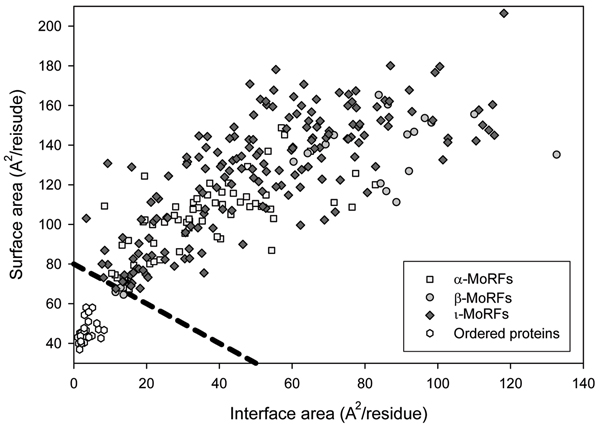
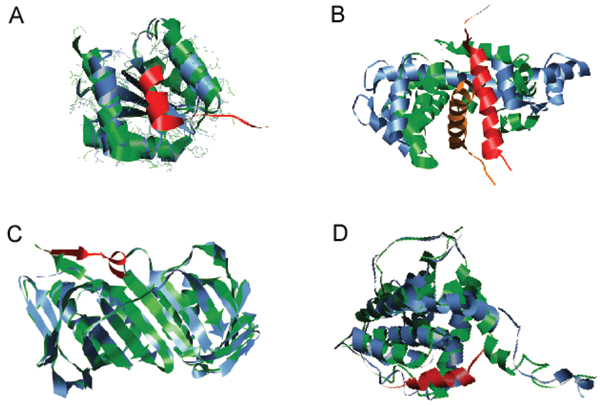
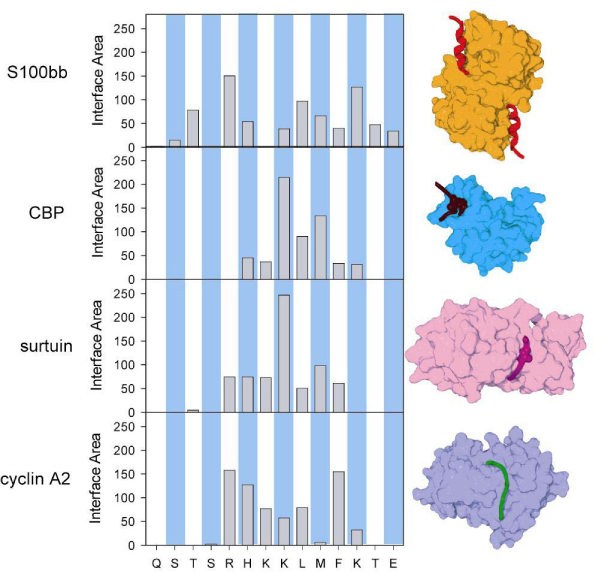
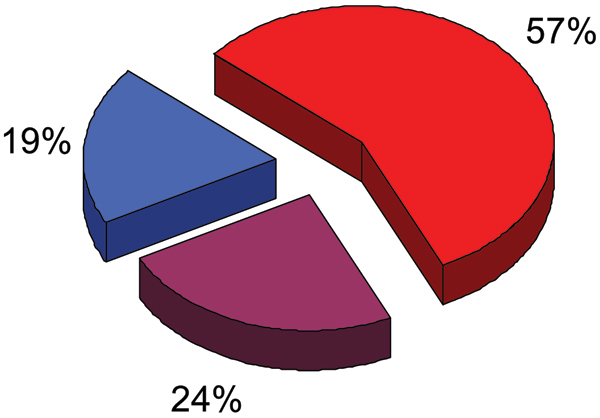
Intrinsically disordered protein is common across the three domains of life, but especially common among the eukaryotic proteomes. Signaling sequences and sites of posttranslational modifications are frequently, or very likely most often, located within regions of intrinsic disorder. Disorder-to-order transitions are coupled with the adoption of different structures with different partners. Also, the flexibility of intrinsic disorder helps different disordered regions to bind to a common binding site on a common partner. Such capacity for binding diversity plays important roles in both protein-protein interaction networks and likely also in gene regulation networks. Such disorder-based signaling is further modulated in multicellular eukaryotes by alternative splicing, for which such splicing events map to regions of disorder much more often than to regions of structure. Associating alternative splicing with disorder rather than structure alleviates theoretical and experimentally observed problems associated with the folding of different length, isomeric amino acid sequences. The combination of disorder and alternative splicing is proposed to provide a mechanism for easily "trying out" different signaling pathways, thereby providing the mechanism for generating signaling diversity and enabling the evolution of cell differentiation and multicellularity. Finally, several recent small molecules of interest as potential drugs have been shown to act by blocking protein-protein interactions based on intrinsic disorder of one of the partners. Study of these examples has led to a new approach for drug discovery, and bioinformatics analysis of the human proteome suggests that various disease-associated proteins are very rich in such disorder-based drug discovery targets.
我们关于蛋白质无序性的首个预测方法于十多年前发表在《IEEE神经网络国际会议论文集》上(罗梅罗P、奥布拉多维奇Z、基辛格C、维拉弗兰卡JE、邓克AK(1997年)从氨基酸序列识别蛋白质中的无序区域。《IEEE神经网络国际会议论文集》,第1卷:90 - 95页)。到目前为止,另外二十多个实验室团队也加入了改进蛋白质无序性预测的工作。虽然用于预测蛋白质内在无序性的各种方法与用于预测二级结构的方法类似,但这两种结构类型完全不同。例如,这两种结构类别具有非常不同的动态特性,不规则二级结构类别比无序类别流动性小得多。二级结构的预测很有用。另一方面,内在无序性的预测具有革命性,导致了对已有一百多年历史的蛋白质结构与功能关系观点的重大修正。几十年来,实验人员一直在提供证据表明,一些蛋白质在生理条件下缺乏固定结构或处于无序(或未折叠)状态。此外,实验人员还表明,对于许多蛋白质来说,它们的功能取决于非结构化而非结构化状态;这样的结果与诸如锁钥假说等有一百多年历史的观点形成了鲜明对比。尽管有关于许多重要例子的广泛数据,包括与疾病相关的蛋白质,但无序性对蛋白质功能的重要性在很大程度上被忽视了。事实上,据我们所知,当前的生物化学书籍甚至都没有给出一个公认的依赖无序性的功能例子,尽管一些关于依赖无序性的功能的报道已有五十多年历史。全基因组内在无序性预测的结果以及其他关于内在无序性的生物信息学研究结果要求人们关注这些蛋白质。
无序性预测对于表明相对较少的经实验表征的例子是大量相关无序蛋白质集合的成员很重要,这些无序蛋白质广泛分布于生命的所有三个域中。现在已知许多重要的生物学功能直接依赖于未折叠或部分折叠状态,或者与之密切相关。在这里,我们的目标是回顾关键发现,并将这些发现整合在一起,以支持理解序列 - 功能关系的新方法。
内在无序蛋白质在生命的三个域中都很常见,但在真核蛋白质组中尤其常见。信号序列和翻译后修饰位点经常,或者很可能最常位于内在无序区域内。无序到有序的转变与采用不同结构与不同伙伴相关联。此外,内在无序性的灵活性有助于不同的无序区域结合到共同伙伴上的一个共同结合位点。这种结合多样性的能力在蛋白质 - 蛋白质相互作用网络中以及可能在基因调控网络中都起着重要作用。在多细胞真核生物中,基于无序性的这种信号传导通过可变剪接进一步调节,对于可变剪接事件来说,映射到无序区域的频率比映射到结构区域的频率高得多。将可变剪接与无序性而非结构联系起来,缓解了与不同长度、异构体氨基酸序列折叠相关的理论和实验观察到的问题。无序性和可变剪接的结合被认为提供了一种机制,用于轻松地“尝试”不同的信号传导途径,从而提供产生信号多样性的机制,并促进细胞分化和多细胞性的进化。最后,最近有几种作为潜在药物备受关注的小分子已被证明通过基于其中一个伙伴的内在无序性来阻断蛋白质 - 蛋白质相互作用而起作用。对这些例子的研究导致了一种新的药物发现方法,对人类蛋白质组的生物信息学分析表明,各种与疾病相关的蛋白质在这种基于无序性的药物发现靶点方面非常丰富。