Wu Jian, Lenchik Nataliya I, Gerling Ivan C
Department of Neurology, Xuan Wu Hospital, Capital Medical University, Beijing, China.
BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S12. doi: 10.1186/1471-2164-9-S2-S12.
As studies of molecular biology system attempt to achieve a comprehensive understanding of a particular system, Type 1 errors may be a significant problem. However, few investigators are inclined to accept the increase in Type 2 errors (false positives) that may result when less stringent statistical cut-off values are used. To address this dilemma, we developed an analysis strategy that used a stringent statistical analysis to create a list of differentially expressed genes that served as "bait" to "fish out" other genes with similar patterns of expression.
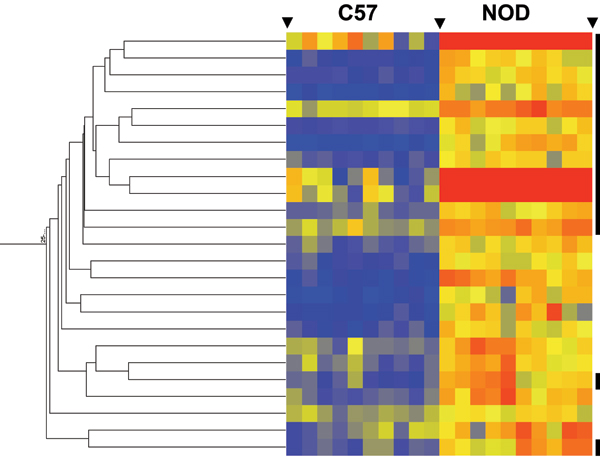
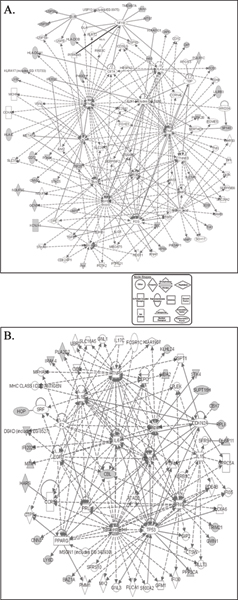
Comparing two strains of mice (NOD and C57Bl/6), we identified 93 genes with statistically significant differences in their patterns of expression. Hierarchical clustering identified an additional 39 genes with similar patterns of expression differences between the two strains. Pathway analysis was then employed: 1) identify the central genes and define biological processes that may be regulated by the genes identified, and 2) identify genes on the lists that could not be connected to each other in pathways (potential false positives). For networks created by both gene lists, the most connected (central) genes were interferon gamma (IFN-gamma) and tumor necrosis factor alpha (TNF-alpha). These two cytokines are relevant to the biological differences between the two strains of mice. Furthermore, the network created by the list of 39 genes also suggested other biological differences between the strains.
Taken together, these data demonstrate how stringent statistical analysis, combined with hierarchical clustering and pathway analysis may offer deeper insight into the biological processes reflected from a set of expression array data. This approach allows us to 'recapture" false negative genes that otherwise would have been missed by the statistical analysis.
随着分子生物学系统研究试图全面了解特定系统,I 型错误可能是一个重大问题。然而,很少有研究者愿意接受在使用不太严格的统计临界值时可能导致的 II 型错误(假阳性)增加。为了解决这一困境,我们开发了一种分析策略,该策略使用严格的统计分析来创建一组差异表达基因列表,这些基因作为“诱饵”来“钓出”具有相似表达模式的其他基因。
比较两种小鼠品系(NOD 和 C57Bl/6),我们鉴定出 93 个基因,其表达模式存在统计学显著差异。层次聚类又鉴定出另外 39 个在两种品系间具有相似表达差异模式的基因。然后进行通路分析:1)识别核心基因并确定可能由所鉴定基因调控的生物学过程,2)识别通路中无法相互连接的列表上的基因(潜在假阳性)。对于由两个基因列表创建的网络,连接性最强(核心)的基因是干扰素γ(IFN-γ)和肿瘤坏死因子α(TNF-α)。这两种细胞因子与两种小鼠品系间的生物学差异相关。此外,由 39 个基因列表创建的网络也表明了品系间的其他生物学差异。
综上所述,这些数据表明严格的统计分析与层次聚类和通路分析相结合如何能更深入洞察一组表达阵列数据所反映的生物学过程。这种方法使我们能够“找回”那些否则会被统计分析遗漏的假阴性基因。