Durot Maxime, Le Fèvre François, de Berardinis Véronique, Kreimeyer Annett, Vallenet David, Combe Cyril, Smidtas Serge, Salanoubat Marcel, Weissenbach Jean, Schachter Vincent
Genoscope (Commissariat à l'Energie Atomique) and UMR 8030 CNRS-Genoscope-Université d'Evry, 2 rue Gaston Crémieux, CP5706, 91057 Evry, Cedex, France.
BMC Syst Biol. 2008 Oct 7;2:85. doi: 10.1186/1752-0509-2-85.
Genome-scale metabolic models are powerful tools to study global properties of metabolic networks. They provide a way to integrate various types of biological information in a single framework, providing a structured representation of available knowledge on the metabolism of the respective species.
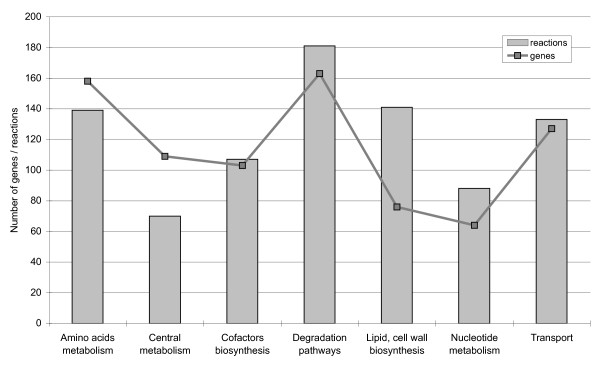
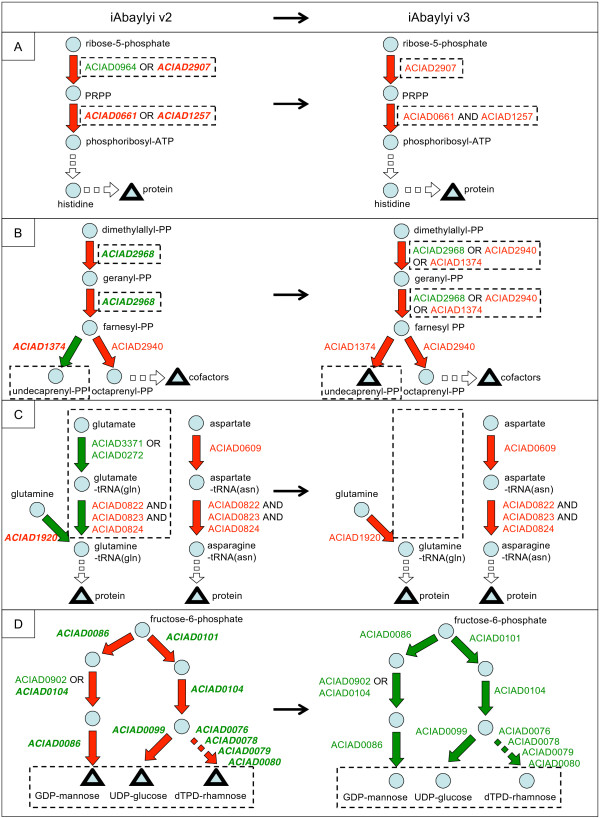
We reconstructed a constraint-based metabolic model of Acinetobacter baylyi ADP1, a soil bacterium of interest for environmental and biotechnological applications with large-spectrum biodegradation capabilities. Following initial reconstruction from genome annotation and the literature, we iteratively refined the model by comparing its predictions with the results of large-scale experiments: (1) high-throughput growth phenotypes of the wild-type strain on 190 distinct environments, (2) genome-wide gene essentialities from a knockout mutant library, and (3) large-scale growth phenotypes of all mutant strains on 8 minimal media. Out of 1412 predictions, 1262 were initially consistent with our experimental observations. Inconsistencies were systematically examined, leading in 65 cases to model corrections. The predictions of the final version of the model, which included three rounds of refinements, are consistent with the experimental results for (1) 91% of the wild-type growth phenotypes, (2) 94% of the gene essentiality results, and (3) 94% of the mutant growth phenotypes. To facilitate the exploitation of the metabolic model, we provide a web interface allowing online predictions and visualization of results on metabolic maps.
The iterative reconstruction procedure led to significant model improvements, showing that genome-wide mutant phenotypes on several media can significantly facilitate the transition from genome annotation to a high-quality model.
基因组规模代谢模型是研究代谢网络全局特性的强大工具。它们提供了一种在单一框架中整合各类生物信息的方式,对相应物种代谢的现有知识进行结构化表示。
我们重建了拜氏不动杆菌ADP1的基于约束的代谢模型,这是一种具有广谱生物降解能力、在环境和生物技术应用方面备受关注的土壤细菌。在根据基因组注释和文献进行初始重建后,我们通过将模型预测结果与大规模实验结果进行比较来迭代优化模型:(1)野生型菌株在190种不同环境下的高通量生长表型,(2)来自基因敲除突变体文库的全基因组基因必需性,以及(3)所有突变体菌株在8种基本培养基上的大规模生长表型。在1412个预测中,1262个最初与我们的实验观察结果一致。我们系统地检查了不一致之处,在65个案例中对模型进行了修正。经过三轮优化的模型最终版本的预测结果与实验结果在以下方面一致:(1)91%的野生型生长表型,(2)94%的基因必需性结果,以及(3)94%的突变体生长表型。为便于利用该代谢模型,我们提供了一个网络界面,允许在线预测并在代谢图上可视化结果。
迭代重建过程使模型得到了显著改进,表明几种培养基上的全基因组突变体表型能够显著促进从基因组注释到高质量模型的转变。