Lecca Davide, Trincavelli Maria Letizia, Gelosa Paolo, Sironi Luigi, Ciana Paolo, Fumagalli Marta, Villa Giovanni, Verderio Claudia, Grumelli Carlotta, Guerrini Uliano, Tremoli Elena, Rosa Patrizia, Cuboni Serena, Martini Claudia, Buffo Annalisa, Cimino Mauro, Abbracchio Maria P
Department of Pharmacological Sciences, University of Milan, Milan, Italy.
PLoS One. 2008;3(10):e3579. doi: 10.1371/journal.pone.0003579. Epub 2008 Oct 31.
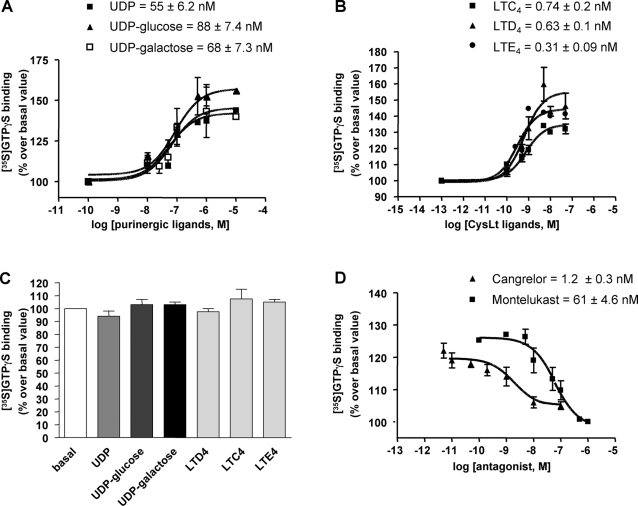
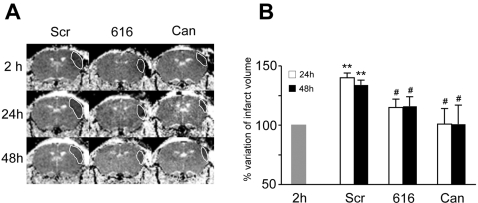
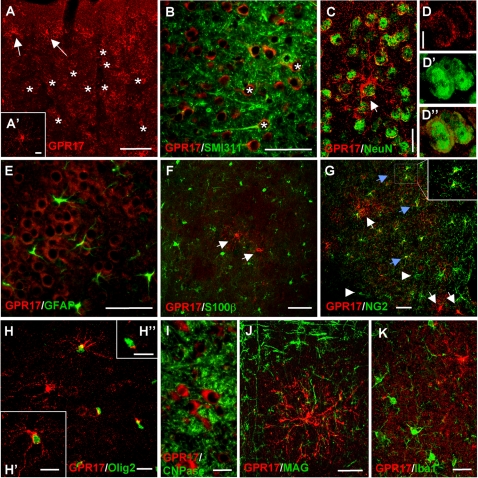
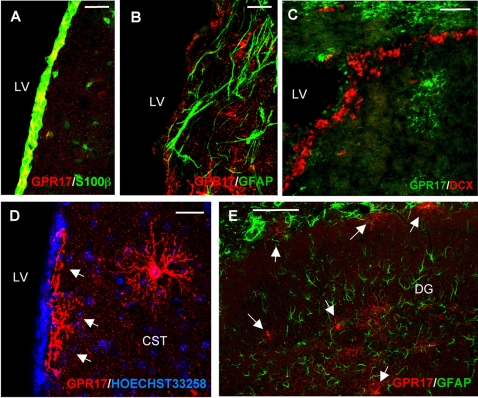
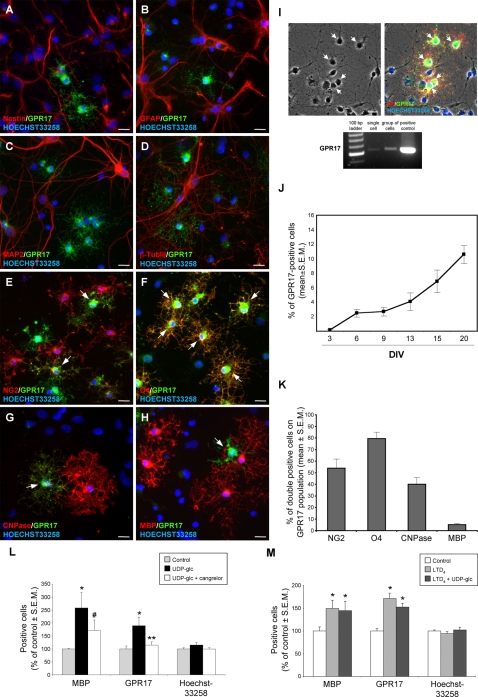
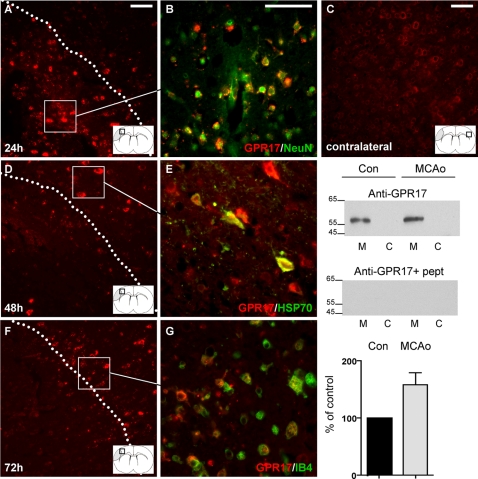
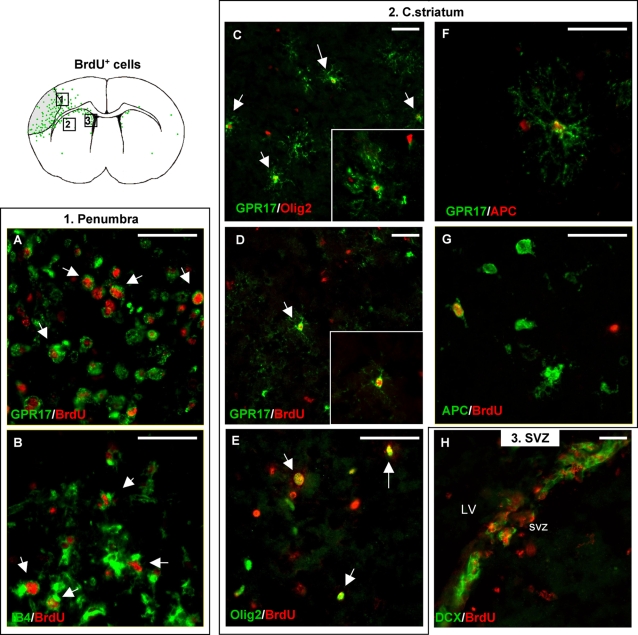
Deciphering the mechanisms regulating the generation of new neurons and new oligodendrocytes, the myelinating cells of the central nervous system, is of paramount importance to address new strategies to replace endogenous damaged cells in the adult brain and foster repair in neurodegenerative diseases. Upon brain injury, the extracellular concentrations of nucleotides and cysteinyl-leukotrienes (cysLTs), two families of endogenous signaling molecules, are markedly increased at the site of damage, suggesting that they may act as "danger signals" to alert responses to tissue damage and start repair. Here we show that, in brain telencephalon, GPR17, a recently deorphanized receptor for both uracil nucleotides and cysLTs (e.g., UDP-glucose and LTD(4)), is normally present on neurons and on a subset of parenchymal quiescent oligodendrocyte precursor cells. We also show that induction of brain injury using an established focal ischemia model in the rodent induces profound spatiotemporal-dependent changes of GPR17. In the lesioned area, we observed an early and transient up-regulation of GPR17 in neurons expressing the cellular stress marker heat shock protein 70. Magnetic Resonance Imaging in living mice showed that the in vivo pharmacological or biotechnological knock down of GPR17 markedly prevents brain infarct evolution, suggesting GPR17 as a mediator of neuronal death at this early ischemic stage. At later times after ischemia, GPR17 immuno-labeling appeared on microglia/macrophages infiltrating the lesioned area to indicate that GPR17 may also acts as a player in the remodeling of brain circuitries by microglia. At this later stage, parenchymal GPR17+ oligodendrocyte progenitors started proliferating in the peri-injured area, suggesting initiation of remyelination. To confirm a specific role for GPR17 in oligodendrocyte differentiation, the in vitro exposure of cortical pre-oligodendrocytes to the GPR17 endogenous ligands UDP-glucose and LTD(4) promoted the expression of myelin basic protein, confirming progression toward mature oligodendrocytes. Thus, GPR17 may act as a "sensor" that is activated upon brain injury on several embryonically distinct cell types, and may play a key role in both inducing neuronal death inside the ischemic core and in orchestrating the local remodeling/repair response. Specifically, we suggest GPR17 as a novel target for therapeutic manipulation to foster repair of demyelinating wounds, the types of lesions that also occur in patients with multiple sclerosis.
阐明调节新神经元和新少突胶质细胞(中枢神经系统的髓鞘形成细胞)生成的机制,对于探寻新策略以替代成体大脑中内源性受损细胞并促进神经退行性疾病的修复至关重要。脑损伤后,两类内源性信号分子——核苷酸和半胱氨酰白三烯(cysLTs)的细胞外浓度在损伤部位显著升高,这表明它们可能作为“危险信号”来警示对组织损伤的反应并启动修复。在此我们表明,在脑端脑中,GPR17(一种最近被确定的尿嘧啶核苷酸和cysLTs——如UDP - 葡萄糖和LTD(4)的受体)通常存在于神经元和实质静止少突胶质细胞前体细胞的一个亚群上。我们还表明,使用已建立的啮齿动物局灶性缺血模型诱导脑损伤会引发GPR17深刻的时空依赖性变化。在损伤区域,我们观察到在表达细胞应激标记热休克蛋白70的神经元中GPR17出现早期短暂上调。活体小鼠的磁共振成像显示,体内对GPR17进行药理学或生物技术敲低可显著阻止脑梗死进展,这表明GPR17在这个早期缺血阶段是神经元死亡的介质。在缺血后的后期,GPR17免疫标记出现在浸润损伤区域的小胶质细胞/巨噬细胞上,这表明GPR17也可能在小胶质细胞对脑回路的重塑中发挥作用。在这个后期阶段,实质GPR17 +少突胶质细胞祖细胞开始在损伤周边区域增殖,提示髓鞘再生开始。为了证实GPR17在少突胶质细胞分化中的特定作用,将皮质前少突胶质细胞在体外暴露于GPR17内源性配体UDP - 葡萄糖和LTD(4)可促进髓鞘碱性蛋白的表达,证实其向成熟少突胶质细胞的进展。因此,GPR17可能作为一种“传感器”,在脑损伤时被多种胚胎来源不同的细胞类型激活,并且可能在诱导缺血核心内的神经元死亡以及协调局部重塑/修复反应中起关键作用。具体而言,我们认为GPR17是促进脱髓鞘损伤修复的治疗操作的一个新靶点,脱髓鞘损伤也是多发性硬化症患者中出现的损伤类型。