Nonyane Bareng A S, Foulkes Andrea S
Division of Biostatistics and Epidemiology, School of Public Health and Health Sciences, University of Massachusetts Amherst, MA, USA.
BMC Genet. 2008 Nov 14;9:71. doi: 10.1186/1471-2156-9-71.
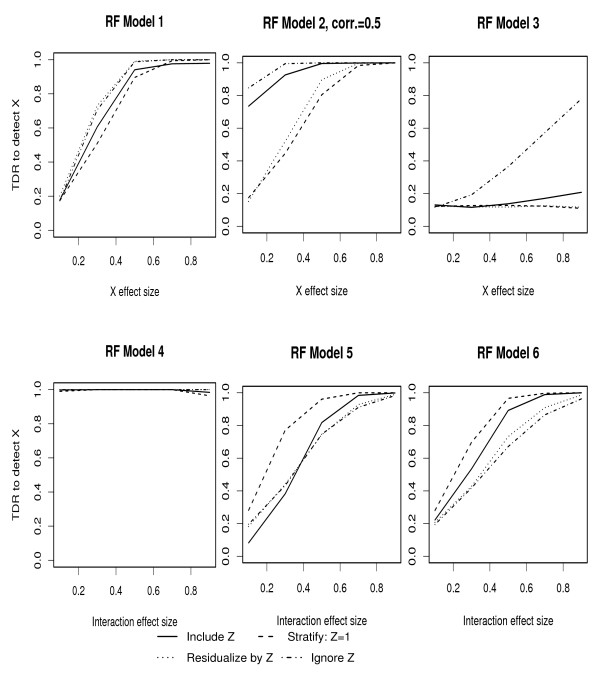
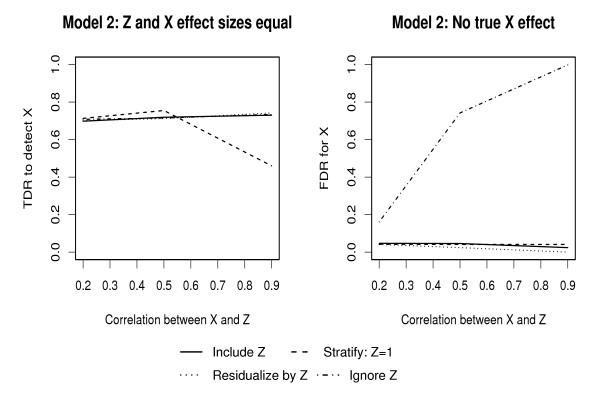
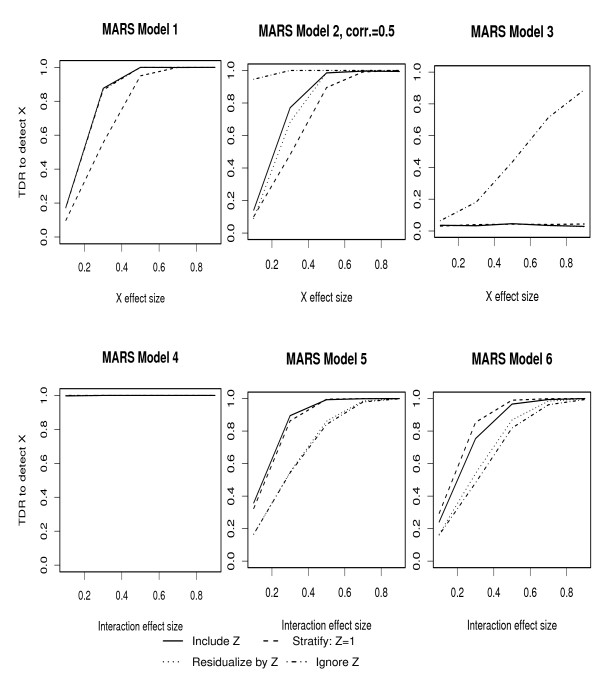
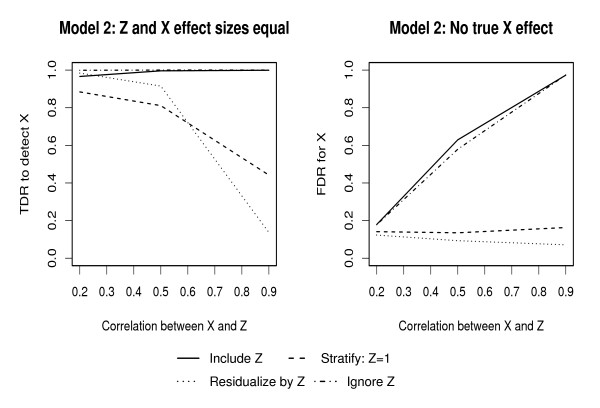
Population-based investigations aimed at uncovering genotype-trait associations often involve high-dimensional genetic polymorphism data as well as information on multiple environmental and clinical parameters. Machine learning (ML) algorithms offer a straightforward analytic approach for selecting subsets of these inputs that are most predictive of a pre-defined trait. The performance of these algorithms, however, in the presence of covariates is not well characterized.
In this manuscript, we investigate two approaches: Random Forests (RFs) and Multivariate Adaptive Regression Splines (MARS). Through multiple simulation studies, the performance under several underlying models is evaluated. An application to a cohort of HIV-1 infected individuals receiving anti-retroviral therapies is also provided.
Consistent with more traditional regression modeling theory, our findings highlight the importance of considering the nature of underlying gene-covariate-trait relationships before applying ML algorithms, particularly when there is potential confounding or effect mediation.
旨在揭示基因型与性状关联的基于人群的调查通常涉及高维基因多态性数据以及多个环境和临床参数的信息。机器学习(ML)算法提供了一种直接的分析方法,用于选择这些输入中对预定义性状最具预测性的子集。然而,这些算法在存在协变量的情况下的性能尚未得到很好的表征。
在本手稿中,我们研究了两种方法:随机森林(RF)和多元自适应回归样条(MARS)。通过多项模拟研究,评估了几种潜在模型下的性能。还提供了对一组接受抗逆转录病毒疗法的HIV-1感染者的应用。
与更传统的回归建模理论一致,我们的研究结果强调了在应用ML算法之前考虑潜在基因-协变量-性状关系性质的重要性,特别是在存在潜在混杂或效应介导的情况下。