BMC Bioinformatics. 2013;14 Suppl 16(Suppl 16):S6. doi: 10.1186/1471-2105-14-S16-S6. Epub 2013 Oct 22.
Multivariate quantitative traits arise naturally in recent neuroimaging genetics studies, in which both structural and functional variability of the human brain is measured non-invasively through techniques such as magnetic resonance imaging (MRI). There is growing interest in detecting genetic variants associated with such multivariate traits, especially in genome-wide studies. Random forests (RFs) classifiers, which are ensembles of decision trees, are amongst the best performing machine learning algorithms and have been successfully employed for the prioritisation of genetic variants in case-control studies. RFs can also be applied to produce gene rankings in association studies with multivariate quantitative traits, and to estimate genetic similarities measures that are predictive of the trait. However, in studies involving hundreds of thousands of SNPs and high-dimensional traits, a very large ensemble of trees must be inferred from the data in order to obtain reliable rankings, which makes the application of these algorithms computationally prohibitive.
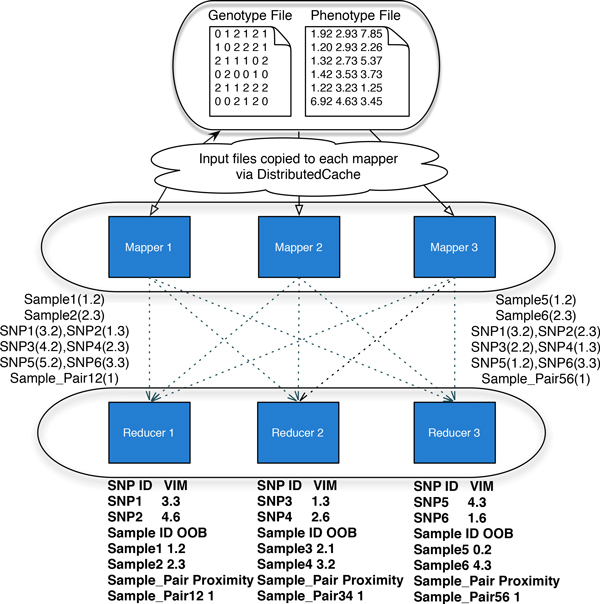
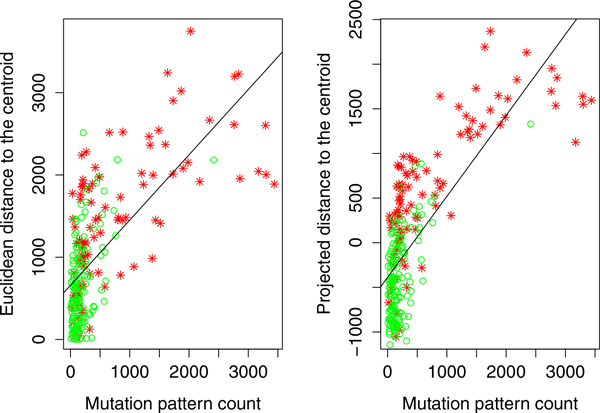
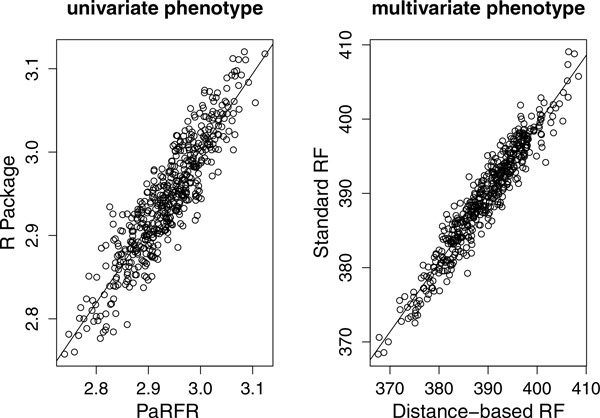
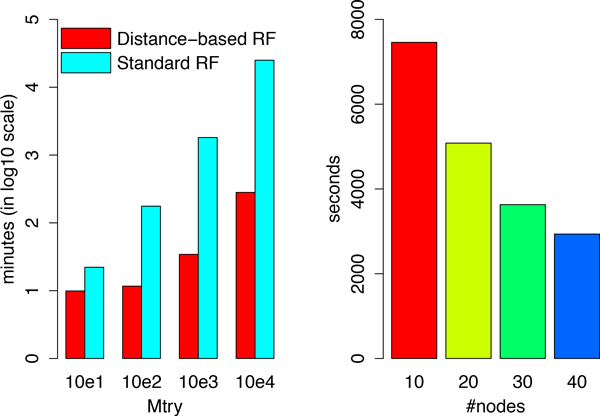
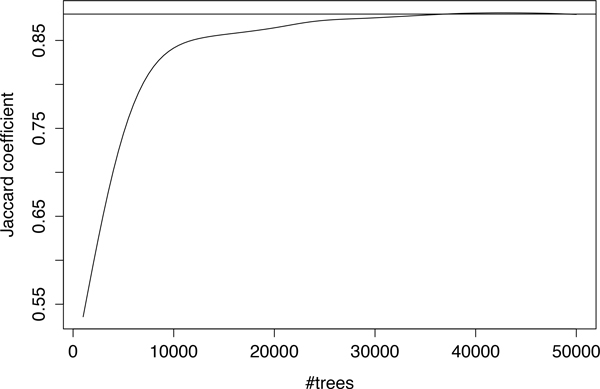
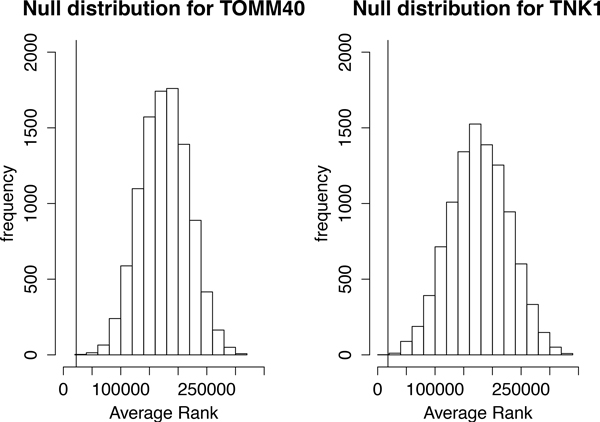
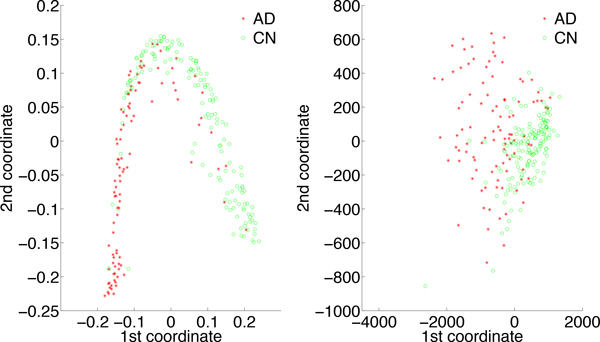
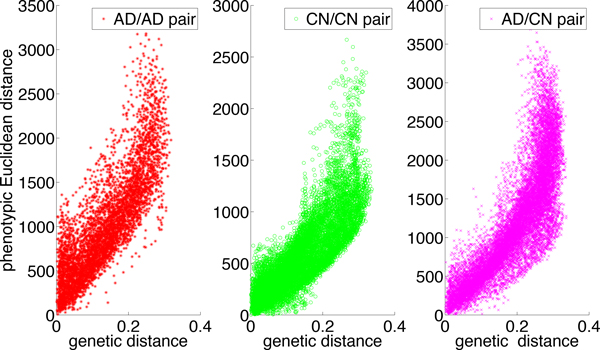
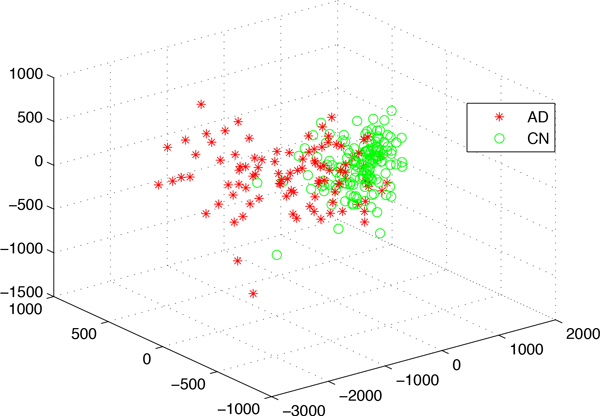
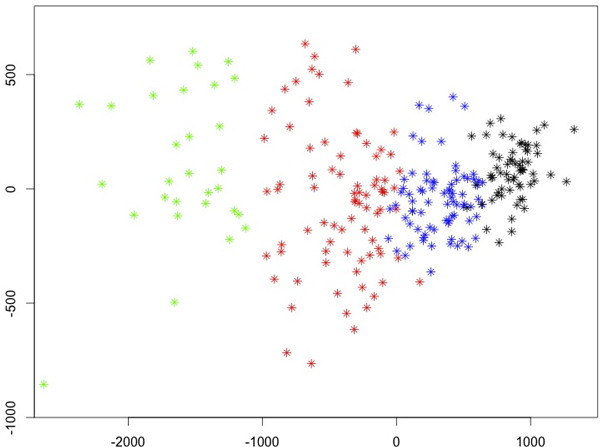
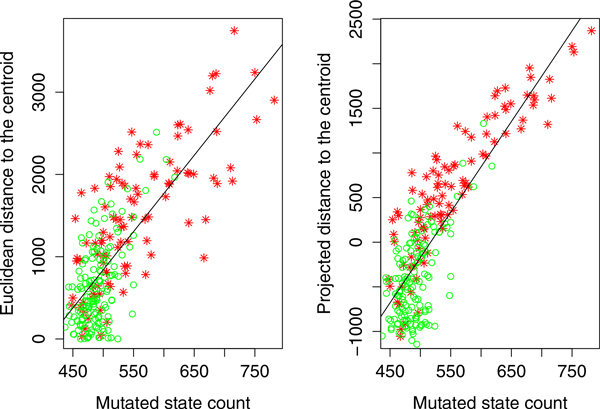
We have developed a parallel version of the RF algorithm for regression and genetic similarity learning tasks in large-scale population genetic association studies involving multivariate traits, called PaRFR (Parallel Random Forest Regression). Our implementation takes advantage of the MapReduce programming model and is deployed on Hadoop, an open-source software framework that supports data-intensive distributed applications. Notable speed-ups are obtained by introducing a distance-based criterion for node splitting in the tree estimation process. PaRFR has been applied to a genome-wide association study on Alzheimer's disease (AD) in which the quantitative trait consists of a high-dimensional neuroimaging phenotype describing longitudinal changes in the human brain structure. PaRFR provides a ranking of SNPs associated to this trait, and produces pair-wise measures of genetic proximity that can be directly compared to pair-wise measures of phenotypic proximity. Several known AD-related variants have been identified, including APOE4 and TOMM40. We also present experimental evidence supporting the hypothesis of a linear relationship between the number of top-ranked mutated states, or frequent mutation patterns, and an indicator of disease severity.
The Java codes are freely available at http://www2.imperial.ac.uk/~gmontana.
多变量数量性状在最近的神经影像学遗传学研究中自然出现,其中通过磁共振成像(MRI)等技术无创地测量人脑的结构和功能变异性。人们越来越感兴趣的是检测与这种多变量性状相关的遗传变异,尤其是在全基因组研究中。随机森林(RF)分类器是决策树的集成,是性能最佳的机器学习算法之一,已成功应用于病例对照研究中遗传变异的优先级排序。RF 还可用于在具有多变量数量性状的关联研究中生成基因排名,并估计遗传相似性度量,这些度量可预测性状。但是,在涉及数十万 SNP 和高维性状的研究中,必须从数据中推断出非常大的树集,以获得可靠的排名,这使得这些算法的应用在计算上变得非常复杂。
我们为涉及多变量性状的大规模人群遗传关联研究中的回归和遗传相似性学习任务开发了 RF 算法的并行版本,称为 PaRFR(并行随机森林回归)。我们的实现利用了 MapReduce 编程模型,并部署在 Hadoop 上,Hadoop 是一个支持数据密集型分布式应用的开源软件框架。通过在树估计过程中引入基于距离的节点分裂标准,可以获得显著的加速。PaRFR 已应用于阿尔茨海默病(AD)的全基因组关联研究,其中数量性状由描述人脑结构纵向变化的高维神经影像学表型组成。PaRFR 提供了与该性状相关的 SNP 排名,并生成了可以直接与表型相似性的成对度量进行比较的遗传相似性的成对度量。已经确定了几个与 AD 相关的变体,包括 APOE4 和 TOMM40。我们还提供了支持突变状态或常见突变模式的数量与疾病严重程度指标之间存在线性关系的实验证据。
Java 代码可在 http://www2.imperial.ac.uk/~gmontana 免费获得。