Kanzaki T, Wang A M, Desnick R J
Department of Dermatology, Nagoya City University Medical School, Japan.
J Clin Invest. 1991 Aug;88(2):707-11. doi: 10.1172/JCI115357.
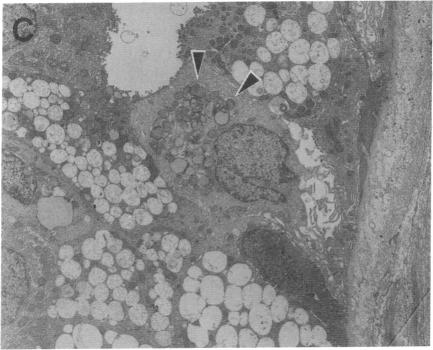
Recently a novel case of angiokeratoma corporis diffusum with glycoaminoaciduria was described in a 46-yr-old Japanese woman. Known causes of the cutaneous manifestation were eliminated by enzyme analyses, and further characterization of the accumulated urinary O-linked sialopeptides revealed identity to those excreted by patients with an infantile neuroaxonal dystrophy due to lysosomal alpha-N-acetylgalactosaminidase deficiency. Investigation of the alpha-N-acetylgalactosaminidase activity and protein in the proband revealed less than 2% of normal activity and the absence of detectable immunoreactive enzyme protein, findings comparable to those in the patients with infantile neuroaxonal dystrophy and alpha-N-acetylgalactosaminidase deficiency. In addition, the proband's unaffected offspring had half-normal levels of alpha-N-acetylgalactosaminidase activity, consistent with this enzymatic deficiency being the primary metabolic defect in this autosomal recessive trait. Ultrastructural examination of skin and blood cells from the adult proband revealed the presence of prominent lysosomal inclusions containing diffuse amorphous and filamentous material. In contrast, these morphologic findings were not observed in the nonneural tissues from patients with infantile neuroaxonal dystrophy and alpha-N-acetylgalactosaminidase deficiency. These studies document the occurrence of two forms of alpha-N-acetylgalactosaminidase deficiency and sialopeptiduria, a severe infantile-onset form of neuroaxonal dystrophy without angiokeratoma or visceral lysosomal inclusions and an adult-onset form characterized by angiokeratoma, extensive lysosomal accumulation of sialoglycopeptides and the absence of detectable neurologic involvement.
最近,一名46岁的日本女性被诊断出患有弥漫性躯体血管角皮瘤合并氨基糖氨酸尿症。通过酶分析排除了已知的皮肤表现病因,进一步对累积的尿O-连接唾液酸肽进行表征,发现其与因溶酶体α-N-乙酰半乳糖胺酶缺乏导致的婴儿神经轴索性营养不良患者所排泄的物质相同。对先证者的α-N-乙酰半乳糖胺酶活性和蛋白质进行研究,发现其活性低于正常水平的2%,且未检测到可免疫反应的酶蛋白,这些发现与婴儿神经轴索性营养不良和α-N-乙酰半乳糖胺酶缺乏患者的情况相似。此外,先证者未受影响的后代α-N-乙酰半乳糖胺酶活性水平为正常的一半,这与这种酶缺乏是该常染色体隐性性状的主要代谢缺陷一致。对成年先证者的皮肤和血细胞进行超微结构检查,发现存在含有弥漫性无定形和丝状物质的显著溶酶体包涵体。相比之下,在婴儿神经轴索性营养不良和α-N-乙酰半乳糖胺酶缺乏患者的非神经组织中未观察到这些形态学发现。这些研究证明了两种形式的α-N-乙酰半乳糖胺酶缺乏和唾液酸肽尿症的存在,一种是严重的婴儿期发病形式的神经轴索性营养不良,无血管角皮瘤或内脏溶酶体包涵体,另一种是成人发病形式,其特征为血管角皮瘤、唾液酸糖肽在溶酶体中广泛积累且无明显神经受累。