Bioinformatics Group, Albert-Ludwigs-University Freiburg, Georges-Koehler-Allee 106, Freiburg, D-79110, Germany.
Bioinformatics. 2009 Aug 15;25(16):2095-102. doi: 10.1093/bioinformatics/btp065. Epub 2009 Feb 2.
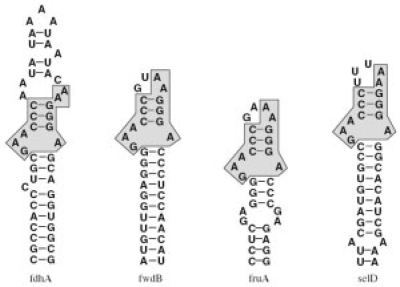
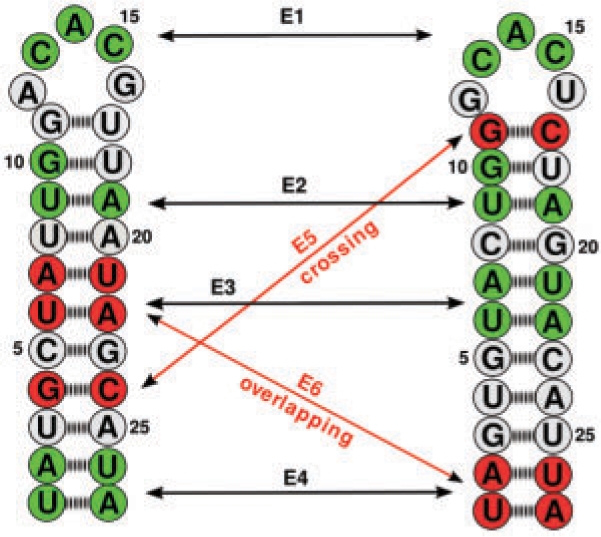
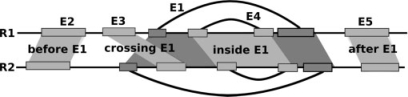
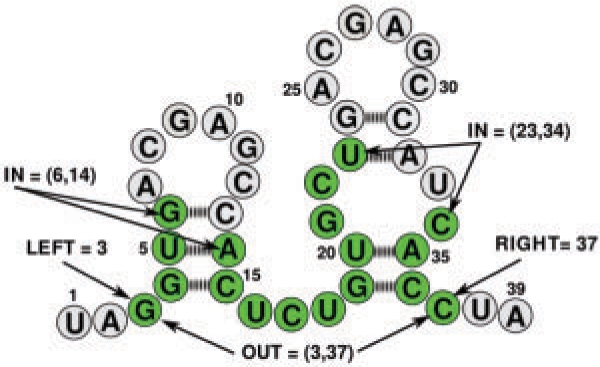
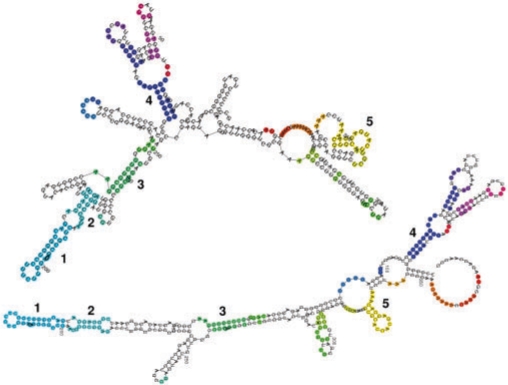
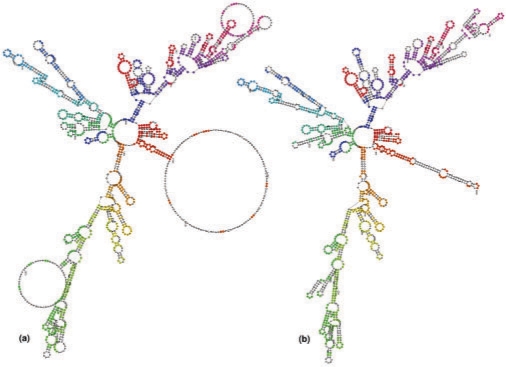
Specific functions of ribonucleic acid (RNA) molecules are often associated with different motifs in the RNA structure. The key feature that forms such an RNA motif is the combination of sequence and structure properties. In this article, we introduce a new RNA sequence-structure comparison method which maintains exact matching substructures. Existing common substructures are treated as whole unit while variability is allowed between such structural motifs. Based on a fast detectable set of overlapping and crossing substructure matches for two nested RNA secondary structures, our method ExpaRNA (exact pattern of alignment of RNA) computes the longest collinear sequence of substructures common to two RNAs in O(H.nm) time and O(nm) space, where H << n.m for real RNA structures. Applied to different RNAs, our method correctly identifies sequence-structure similarities between two RNAs.
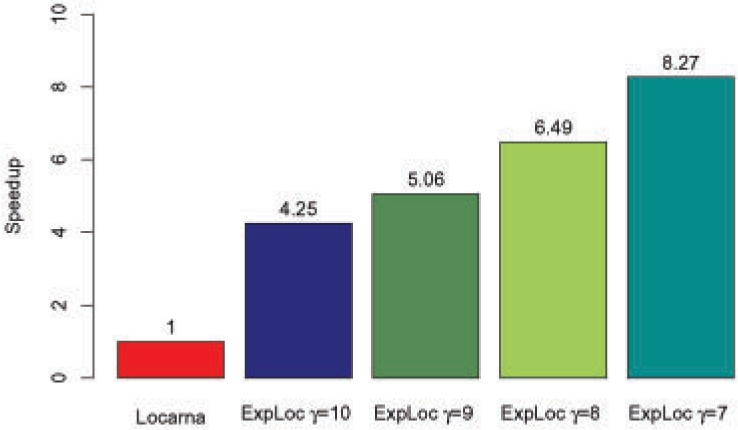
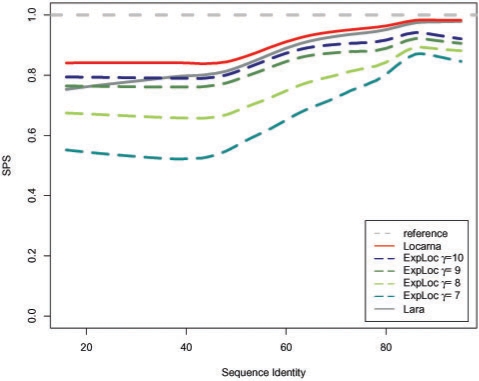
We have compared ExpaRNA with two other alignment methods that work with given RNA structures, namely RNAforester and RNA_align. The results are in good agreement, but can be obtained in a fraction of running time, in particular for larger RNAs. We have also used ExpaRNA to speed up state-of-the-art Sankoff-style alignment tools like LocARNA, and observe a tradeoff between quality and speed. However, we get a speedup of 4.25 even in the highest quality setting, where the quality of the produced alignment is comparable to that of LocARNA alone.
The presented algorithm is implemented in the program ExpaRNA, which is available from our website (http://www.bioinf.uni-freiburg.de/Software).
核糖核酸(RNA)分子的特定功能通常与 RNA 结构中的不同基序相关联。形成这种 RNA 基序的关键特征是序列和结构特性的组合。在本文中,我们引入了一种新的 RNA 序列-结构比较方法,该方法保持精确匹配的子结构。现有的常见子结构被视为整体单元,同时允许这些结构基序之间存在可变性。基于两个嵌套 RNA 二级结构的可检测重叠和交叉子结构匹配的快速检测集,我们的方法 ExpaRNA(RNA 对齐的精确模式)在 O(H.nm)时间和 O(nm)空间中计算两个 RNA 之间共有子结构的最长共线性序列,其中 H << n.m 适用于真实的 RNA 结构。应用于不同的 RNA,我们的方法正确识别两个 RNA 之间的序列-结构相似性。
我们将 ExpaRNA 与另外两种适用于给定 RNA 结构的对齐方法 RNAforester 和 RNA_align 进行了比较。结果非常吻合,但运行时间可以缩短到一小部分,特别是对于较大的 RNA。我们还使用 ExpaRNA 来加速 Sankoff 风格的对齐工具,如 LocARNA,并观察到质量和速度之间的权衡。然而,即使在最高质量设置下,我们也可以获得 4.25 的加速,其中产生的对齐质量与单独使用 LocARNA 的质量相当。
所提出的算法在程序 ExpaRNA 中实现,该程序可从我们的网站(http://www.bioinf.uni-freiburg.de/Software)获得。