Center for Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Evol Bioinform Online. 2008 Jun 18;4:217-23. doi: 10.4137/ebo.s863.
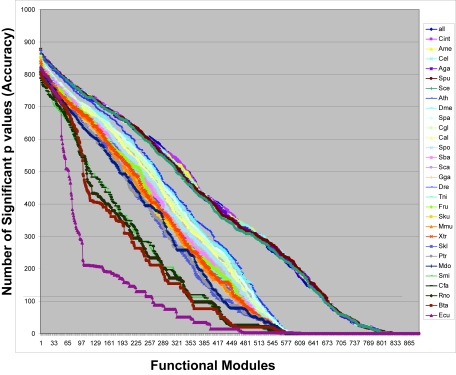
A phylogenetic profile captures the pattern of gene gain and loss throughout evolutionary time. Proteins that interact directly or indirectly within the cell to perform a biological function will often co-evolve, and this co-evolution should be well reflected within their phylogenetic profiles. Thus similar phylogenetic profiles are commonly used for grouping proteins into functional groups. However, it remains unclear how the size and content of the phylogenetic profile impacts the ability to predict function, particularly in Eukaryotes. Here we developed a straightforward approach to address this question by constructing a complete set of phylogenetic profiles for 31 fully sequenced Eukaryotes. Using Gene Ontology as our gold standard, we compared the accuracy of functional predictions made by a comprehensive array of permutations on the complete set of genomes. Our permutations showed that phylogenetic profiles containing between 25 and 31 Eukaryotic genomes performed equally well and significantly better than all other permuted genome sets, with one exception: we uncovered a core of group of 18 genomes that achieved statistically identical accuracy. This core group contained genomes from each branch of the eukaryotic phylogeny, but also contained several groups of closely related organisms, suggesting that a balance between phylogenetic breadth and depth may improve our ability to use Eukaryotic specific phylogenetic profiles for functional annotations.
系统发生谱捕获了整个进化过程中基因获得和丢失的模式。在细胞内直接或间接相互作用以执行生物功能的蛋白质通常会共同进化,并且这种共同进化应该在它们的系统发生谱中得到很好的反映。因此,相似的系统发生谱通常用于将蛋白质分组为功能组。然而,系统发生谱的大小和内容如何影响功能预测的能力仍不清楚,尤其是在真核生物中。在这里,我们通过构建 31 个完全测序的真核生物的完整系统发生谱集,开发了一种简单的方法来解决这个问题。使用基因本体论作为我们的黄金标准,我们比较了在完整基因组集上进行的各种排列的功能预测的准确性。我们的排列表明,包含 25 到 31 个真核生物基因组的系统发生谱表现同样出色,并且显著优于所有其他排列的基因组集,只有一个例外:我们发现了一组由 18 个基因组组成的核心,其准确性达到了统计学上的相同水平。这个核心组包含了真核生物进化枝的每个分支的基因组,但也包含了几组密切相关的生物体,这表明在系统发生广度和深度之间取得平衡可能会提高我们使用真核生物特有的系统发生谱进行功能注释的能力。