Laboratoire de Biologie et Génomique Intégrative, Département de Biologie et Génomique Structurales, Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS/INSERM/UDS, Illkirch, France.
BMC Bioinformatics. 2009 Nov 24;10:383. doi: 10.1186/1471-2105-10-383.
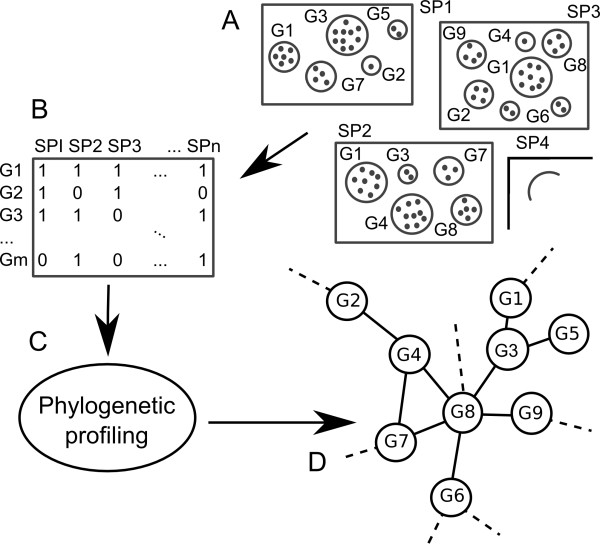
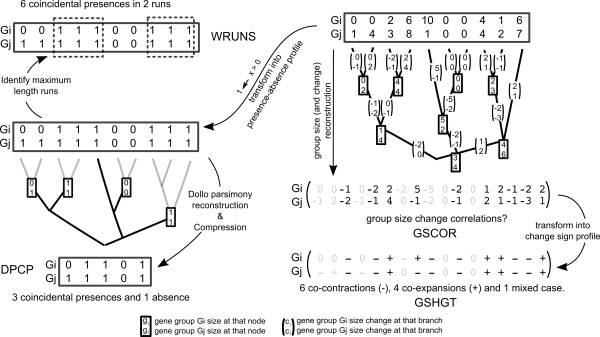
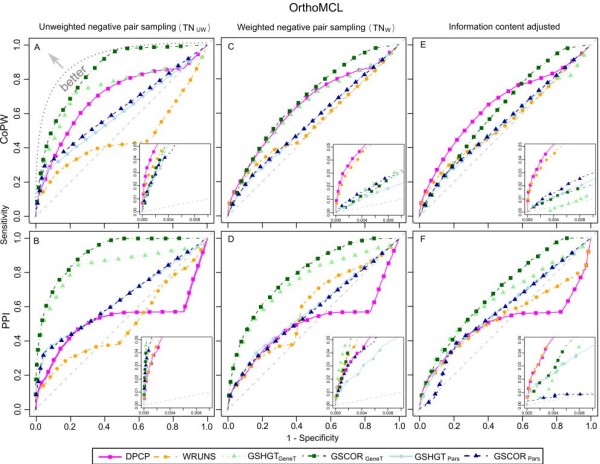
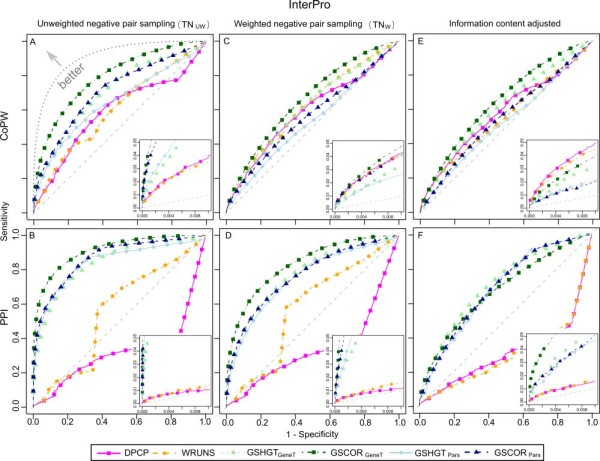
Phylogenetic profiling encompasses an important set of methodologies for in silico high throughput inference of functional relationships between genes. The simplest profiles represent the distribution of gene presence-absence in a set of species as a sequence of 0's and 1's, and it is assumed that functionally related genes will have more similar profiles. The methodology has been successfully used in numerous studies of prokaryotic genomes, although its application in eukaryotes appears problematic, with reported low accuracy due to the complex genomic organization within this domain of life. Recently some groups have proposed an alternative approach based on the correlation of homologous gene group sizes, taking into account all potentially informative genetic events leading to a change in group size, regardless of whether they result in a de novo group gain or total gene group loss.
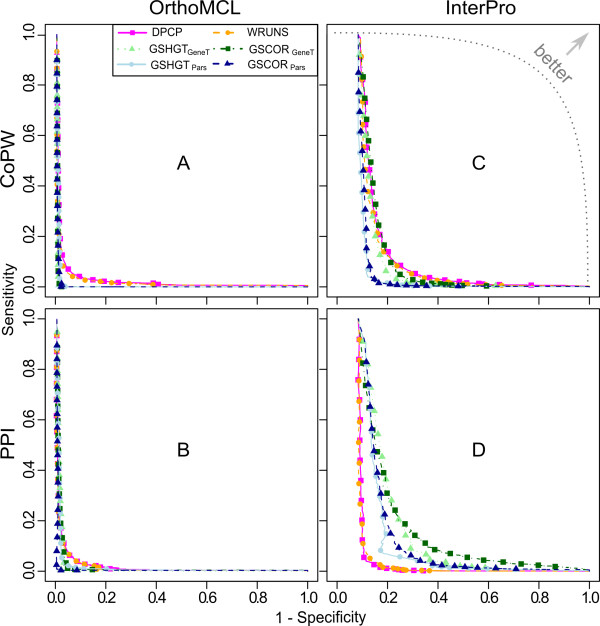
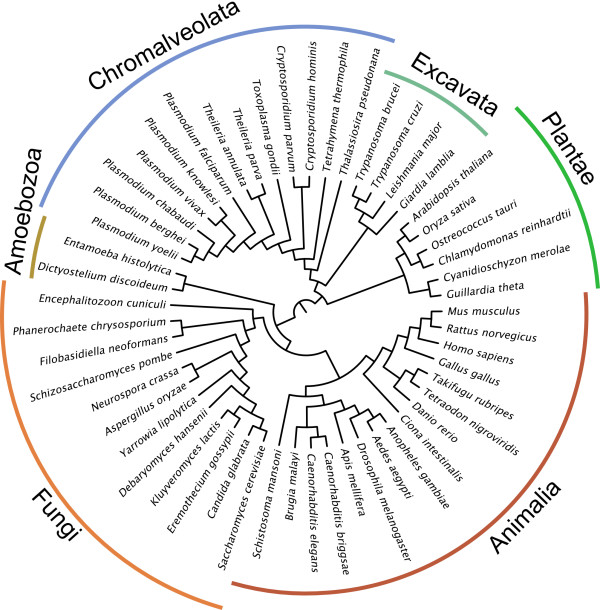
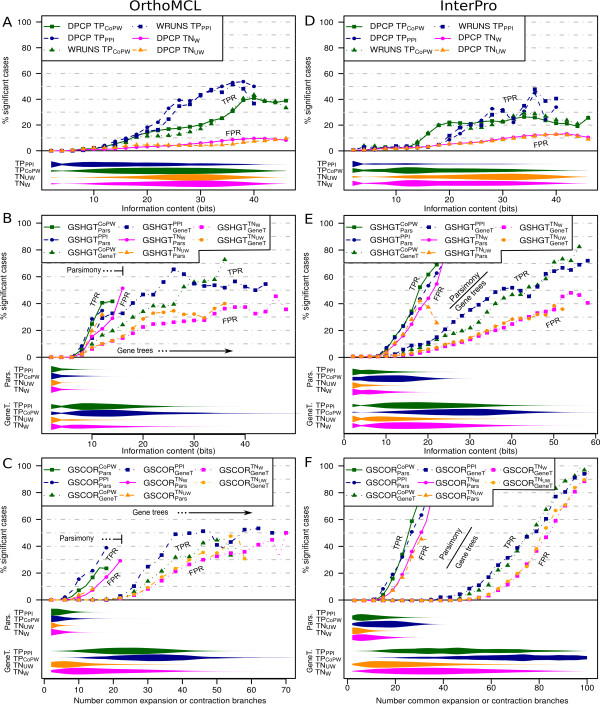
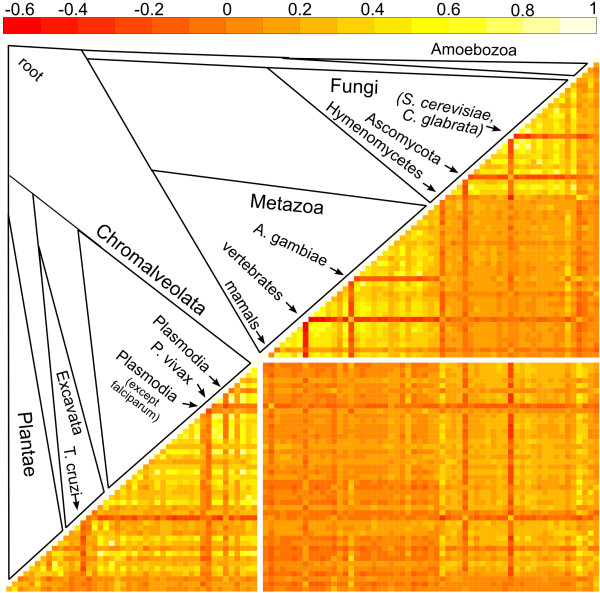
We have compared the performance of classical presence-absence and group size based approaches using a large, diverse set of eukaryotic species. In contrast to most previous comparisons in Eukarya, we take into account the species phylogeny. We also compare the approaches using two different group categories, based on orthology and on domain-sharing. Our results confirm a limited overall performance of phylogenetic profiling in eukaryotes. Although group size based approaches initially showed an increase in performance for the domain-sharing based groups, this seems to be an overestimation due to a simplistic negative control dataset and the choice of null hypothesis rejection criteria.
Presence-absence profiling represents a more accurate classifier of related versus non-related profile pairs, when the profiles under consideration have enough information content. Group size based approaches provide a complementary means of detecting domain or family level co-evolution between groups that may be elusive to presence-absence profiling. Moreover positive correlation between co-evolution scores and functional links imply that these methods could be used to estimate functional distances between gene groups and to cluster them based on their functional relatedness. This study should have important implications for the future development and application of phylogenetic profiling methods, not only in eukaryotic, but also in prokaryotic datasets.
系统发育分析涵盖了一组重要的方法,用于通过计算机对基因之间的功能关系进行高通量推断。最简单的分析方法是将一组物种中的基因存在或缺失情况表示为 0 和 1 的序列,并且假设功能相关的基因将具有更相似的分析方法。该方法已成功应用于许多原核基因组的研究中,尽管由于生命领域内的复杂基因组组织,其在真核生物中的应用似乎存在问题,报告的准确性较低。最近,一些小组提出了一种基于同源基因组大小相关性的替代方法,考虑了导致基因组大小变化的所有潜在信息遗传事件,无论它们是否导致新的基因组获得或基因组完全丢失。
我们使用大量不同的真核生物物种比较了经典的存在缺失和基因组大小分析方法的性能。与真核生物中大多数先前的比较不同,我们考虑了物种的系统发育关系。我们还使用基于同源性和基于结构域共享的两种不同的基因组类别来比较这些方法。我们的结果证实了系统发育分析在真核生物中的整体性能有限。尽管基于基因组大小的方法最初在基于结构域共享的基因组中显示出性能的提高,但由于简单的负控制数据集和选择的无效假设拒绝标准,这似乎是一种高估。
当考虑的分析方法具有足够的信息量时,基于存在缺失的分析方法是一种更准确的相关与非相关分析方法。基于基因组大小的方法提供了一种互补的方法,用于检测基因组之间的域或家族水平的共同进化,这可能是基于存在缺失的分析方法难以检测到的。此外,共同进化评分与功能链接之间的正相关关系意味着,这些方法可用于估计基因组之间的功能距离,并根据其功能相关性对它们进行聚类。这项研究对于未来系统发育分析方法的发展和应用具有重要意义,不仅在真核生物中,而且在原核生物数据集中也是如此。