Petrie Kirsten A, Lee Wen Hwa, Bullock Alex N, Pointon Jenny J, Smith Roger, Russell R Graham G, Brown Matthew A, Wordsworth B Paul, Triffitt James T
Institute of Musculoskeletal Sciences, Botnar Research Centre, Nuffield Department of Orthopaedic Surgery, University of Oxford, Oxford, United Kingdom.
PLoS One. 2009;4(3):e5005. doi: 10.1371/journal.pone.0005005. Epub 2009 Mar 30.
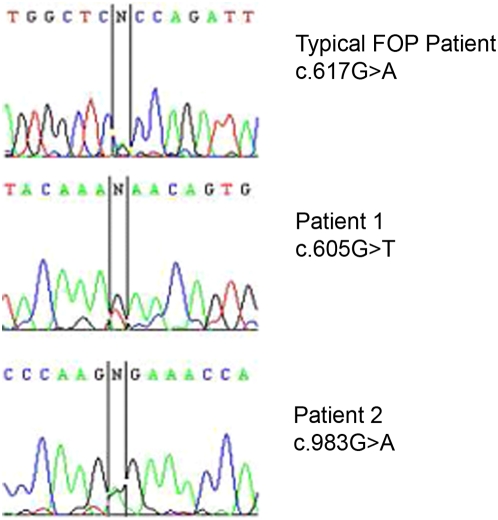
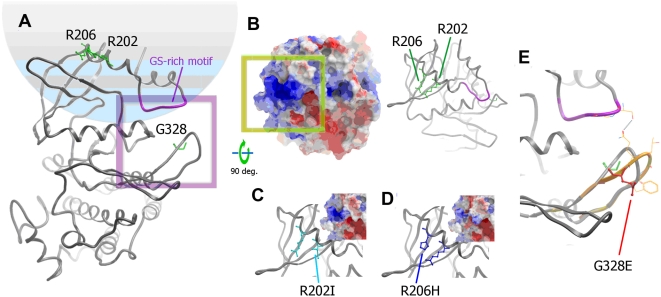
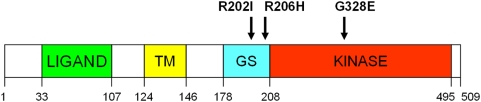
Fibrodysplasia Ossificans Progressiva (FOP) is a rare, heritable condition typified by progression of extensive ossification within skeletal muscle, ligament and tendon together with defects in skeletal development. The condition is easily diagnosed by the presence of shortened great toes and there is severe advancement of disability with age. FOP has been shown to result from a point mutation (c.617G>A) in the ACVR1 gene in almost all patients reported. Very recently two other mutations have been described in three FOP patients. We present here evidence for two further unique mutations (c.605G>T and c.983G>A) in this gene in two FOP patients with some atypical digit abnormalities and other clinical features. The observation of disparate missense mutations mapped to the GS and kinase domains of the protein supports the disease model of mild kinase activation and provides a potential rationale for phenotypic variation.
进行性骨化性纤维发育不良(FOP)是一种罕见的遗传性疾病,其典型特征是骨骼肌、韧带和肌腱内广泛骨化的进展以及骨骼发育缺陷。通过大脚趾缩短可轻易诊断出该疾病,并且随着年龄增长,残疾程度会严重加重。几乎所有已报道的患者中,FOP已被证明是由ACVR1基因中的一个点突变(c.617G>A)引起的。最近,在三名FOP患者中又描述了另外两种突变。我们在此展示了两名具有一些非典型手指异常和其他临床特征的FOP患者中该基因的另外两种独特突变(c.605G>T和c.983G>A)的证据。映射到该蛋白质的GS和激酶结构域的不同错义突变的观察结果支持了轻度激酶激活的疾病模型,并为表型变异提供了潜在的理论依据。