Zhou Leming, Pertea Mihaela, Delcher Arthur L, Florea Liliana
Department of Computer Science, George Washington University, Washington, DC 20052, USA.
Nucleic Acids Res. 2009 Jun;37(11):e80. doi: 10.1093/nar/gkp319. Epub 2009 May 8.
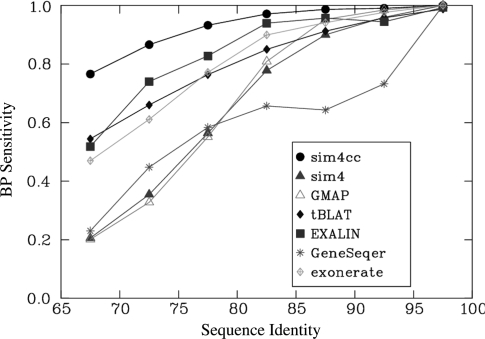
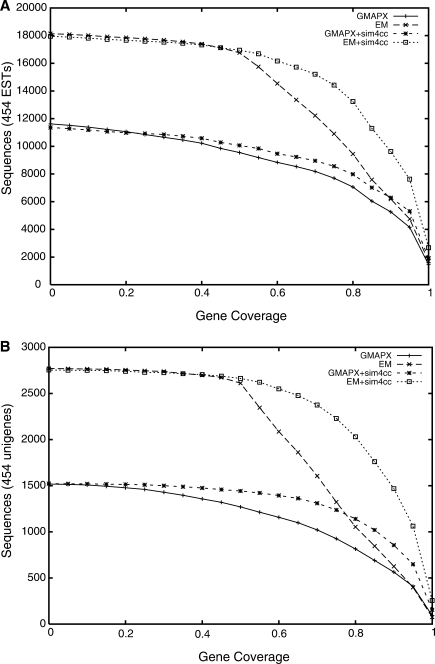
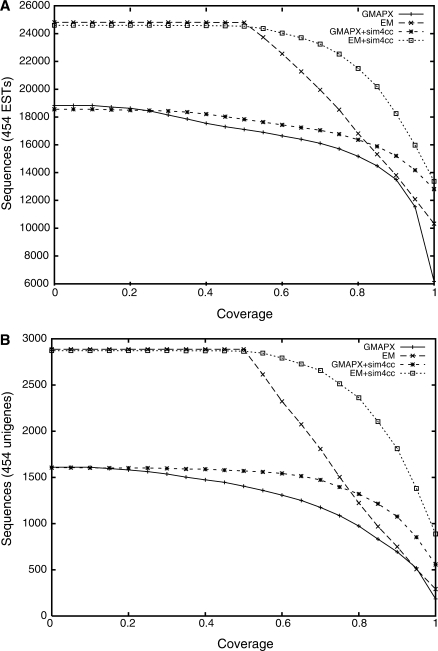
Advances in sequencing technologies have accelerated the sequencing of new genomes, far outpacing the generation of gene and protein resources needed to annotate them. Direct comparison and alignment of existing cDNA sequences from a related species is an effective and readily available means to determine genes in the new genomes. Current spliced alignment programs are inadequate for comparing sequences between different species, owing to their low sensitivity and splice junction accuracy. A new spliced alignment tool, sim4cc, overcomes problems in the earlier tools by incorporating three new features: universal spaced seeds, to increase sensitivity and allow comparisons between species at various evolutionary distances, and powerful splice signal models and evolutionarily-aware alignment techniques, to improve the accuracy of gene models. When tested on vertebrate comparisons at diverse evolutionary distances, sim4cc had significantly higher sensitivity compared to existing alignment programs, more than 10% higher than the closest competitor for some comparisons, while being comparable in speed to its predecessor, sim4. Sim4cc can be used in one-to-one or one-to-many comparisons of genomic and cDNA sequences, and can also be effectively incorporated into a high-throughput annotation engine, as demonstrated by the mapping of 64,000 Fagus grandifolia 454 ESTs and unigenes to the poplar genome.
测序技术的进步加速了新基因组的测序,其速度远远超过注释这些基因组所需的基因和蛋白质资源的生成速度。直接比较和比对来自相关物种的现有cDNA序列是确定新基因组中基因的一种有效且易于获得的方法。由于当前的剪接比对程序灵敏度低且剪接位点准确性差,因此不足以用于比较不同物种之间的序列。一种新的剪接比对工具sim4cc通过融入三个新特性克服了早期工具存在的问题:通用间隔种子,用于提高灵敏度并允许比较不同进化距离的物种;强大的剪接信号模型和具有进化意识的比对技术,用于提高基因模型的准确性。在对不同进化距离的脊椎动物进行比较测试时,与现有的比对程序相比,sim4cc具有显著更高的灵敏度,在某些比较中比最接近的竞争对手高出10%以上,而其速度与前身sim4相当。Sim4cc可用于基因组序列和cDNA序列的一对一或一对多比较,并且还可以有效地整合到高通量注释引擎中,如将64,000个大叶水青冈454 EST和单基因定位到杨树基因组所证明的那样。