Zaslavskiy Mikhail, Bach Francis, Vert Jean-Philippe
Centre for Computational Biology, Mines ParisTech, 35 rue Saint-Honoré, Fontainebleau, Institut Curie, INSERM, U900, Paris, France.
Bioinformatics. 2009 Jun 15;25(12):i259-67. doi: 10.1093/bioinformatics/btp196.
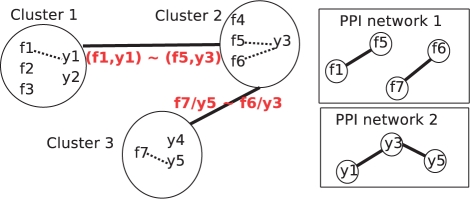
Aligning protein-protein interaction (PPI) networks of different species has drawn a considerable interest recently. This problem is important to investigate evolutionary conserved pathways or protein complexes across species, and to help in the identification of functional orthologs through the detection of conserved interactions. It is, however, a difficult combinatorial problem, for which only heuristic methods have been proposed so far.
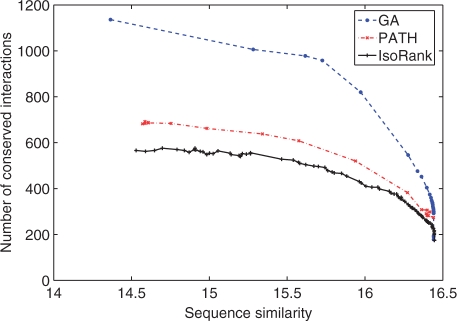
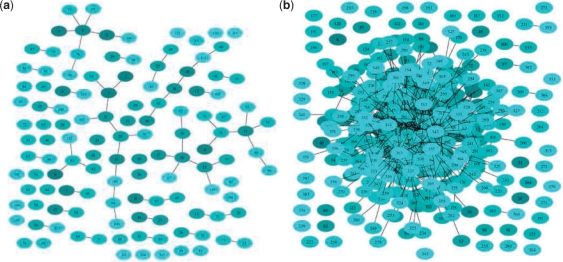
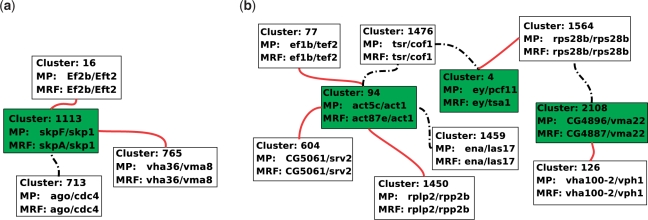
We reformulate the PPI alignment as a graph matching problem, and investigate how state-of-the-art graph matching algorithms can be used for that purpose. We differentiate between two alignment problems, depending on whether strict constraints on protein matches are given, based on sequence similarity, or whether the goal is instead to find an optimal compromise between sequence similarity and interaction conservation in the alignment. We propose new methods for both cases, and assess their performance on the alignment of the yeast and fly PPI networks. The new methods consistently outperform state-of-the-art algorithms, retrieving in particular 78% more conserved interactions than IsoRank for a given level of sequence similarity.
All data and codes are freely and publicly available upon request.
不同物种蛋白质-蛋白质相互作用(PPI)网络的比对近来引起了广泛关注。该问题对于研究跨物种的进化保守途径或蛋白质复合物,以及通过检测保守相互作用来帮助识别功能直系同源物具有重要意义。然而,这是一个困难的组合问题,目前仅提出了启发式方法。
我们将PPI比对重新表述为一个图匹配问题,并研究如何将最先进的图匹配算法用于此目的。根据是否基于序列相似性对蛋白质匹配给出严格约束,或者目标是否是在比对中找到序列相似性和相互作用保守性之间的最佳折衷,我们区分了两种比对问题。我们针对这两种情况都提出了新方法,并在酵母和果蝇PPI网络的比对上评估了它们的性能。新方法始终优于最先进的算法,在给定的序列相似性水平下,与IsoRank相比,尤其能多检索出78%的保守相互作用。
所有数据和代码可根据要求免费公开获取。