Fernández Luis, Nevado Julián, Santos Fernando, Heine-Suñer Damià, Martinez-Glez Victor, García-Miñaur Sixto, Palomo Rebeca, Delicado Alicia, Pajares Isidora López, Palomares María, García-Guereta Luis, Valverde Eva, Hawkins Federico, Lapunzina Pablo
Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, Madrid, Spain.
BMC Med Genet. 2009 Jun 2;10:48. doi: 10.1186/1471-2350-10-48.
Individuals affected with DiGeorge and Velocardiofacial syndromes present with both phenotypic diversity and variable expressivity. The most frequent clinical features include conotruncal congenital heart defects, velopharyngeal insufficiency, hypocalcemia and a characteristic craniofacial dysmorphism. The etiology in most patients is a 3 Mb recurrent deletion in region 22q11.2. However, cases of infrequent deletions and duplications with different sizes and locations have also been reported, generally with a milder, slightly different phenotype for duplications but with no clear genotype-phenotype correlation to date.
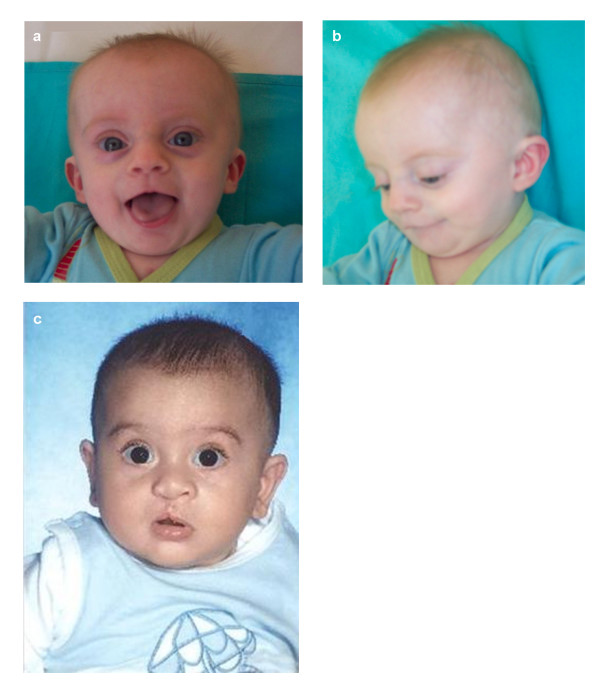
We present a 7 month-old male patient with surgically corrected ASD and multiple VSDs, and dysmorphic facial features not clearly suggestive of 22q11.2 deletion syndrome, and a newborn male infant with cleft lip and palate and upslanting palpebral fissures. Karyotype, FISH, MLPA, microsatellite markers segregation studies and SNP genotyping by array-CGH were performed in both patients and parents.
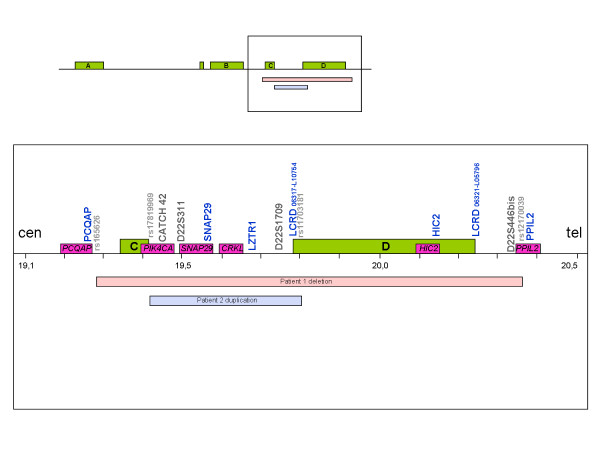
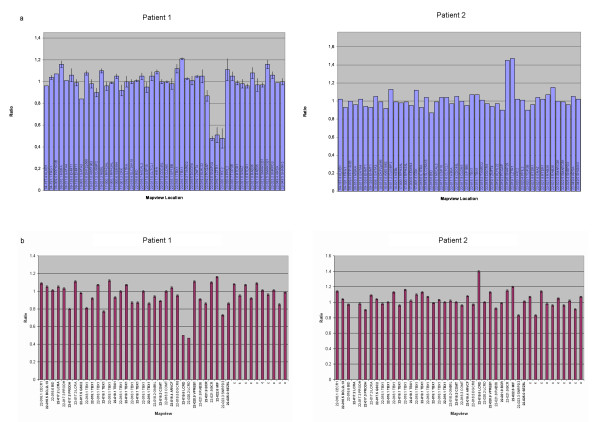
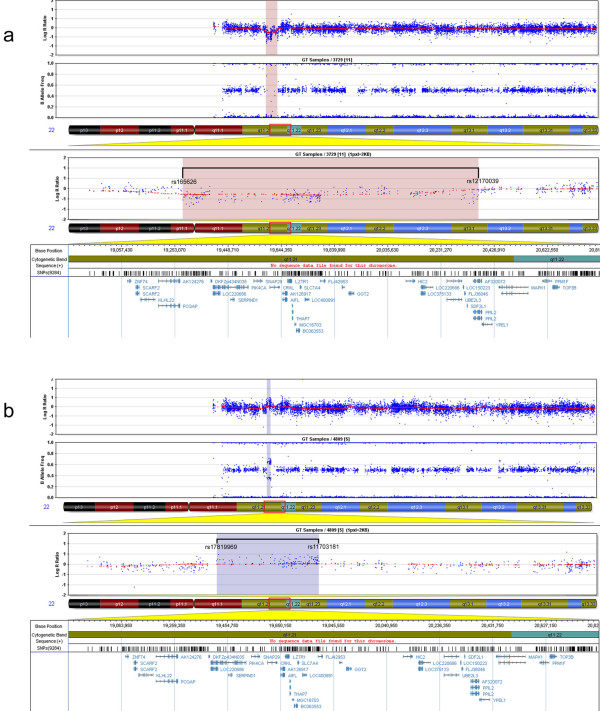
Karyotype and FISH with probe N25 were normal for both patients. MLPA analysis detected a partial de novo 1.1 Mb deletion in one patient and a novel partial familial 0.4 Mb duplication in the other. Both of these alterations were located at a distal position within the commonly deleted region in 22q11.2. These rearrangements were confirmed and accurately characterized by microsatellite marker segregation studies and SNP array genotyping.
The phenotypic diversity found for deletions and duplications supports a lack of genotype-phenotype correlation in the vicinity of the LCRC-LCRD interval of the 22q11.2 chromosomal region, whereas the high presence of duplications in normal individuals supports their role as polymorphisms. We suggest that any hypothetical correlation between the clinical phenotype and the size and location of these alterations may be masked by other genetic and/or epigenetic modifying factors.
患有迪乔治综合征和心脏颜面综合征的个体表现出表型多样性和可变表达性。最常见的临床特征包括圆锥动脉干先天性心脏缺陷、腭咽功能不全、低钙血症和特征性颅面畸形。大多数患者的病因是22q11.2区域的3 Mb重复缺失。然而,也有报道称存在大小和位置不同的罕见缺失和重复病例,重复病例的表型通常较轻且略有不同,但迄今为止尚无明确的基因型-表型相关性。
我们报告了一名7个月大的男性患者,患有经手术矫正的房间隔缺损和多个室间隔缺损,面部畸形特征不明确提示22q11.2缺失综合征,以及一名患有唇腭裂和上斜睑裂的新生儿男婴。对两名患者及其父母进行了核型分析、荧光原位杂交(FISH)、多重连接依赖探针扩增(MLPA)、微卫星标记分离研究以及通过比较基因组杂交芯片进行单核苷酸多态性(SNP)基因分型。
两名患者的核型分析以及使用探针N25的FISH结果均正常。MLPA分析在一名患者中检测到一个1.1 Mb的部分新发缺失,在另一名患者中检测到一个新的0.4 Mb的部分家族性重复。这两种改变均位于22q11.2常见缺失区域的远端位置。这些重排通过微卫星标记分离研究和SNP芯片基因分型得到证实并准确表征。
缺失和重复所发现的表型多样性支持22q11.2染色体区域LCRC-LCRD区间附近缺乏基因型-表型相关性,而正常个体中重复的高发生率支持其作为多态性的作用。我们认为,这些改变的临床表型与大小和位置之间的任何假设相关性可能被其他遗传和/或表观遗传修饰因素所掩盖。