Petersen Malte, Meusemann Karen, Donath Alexander, Dowling Daniel, Liu Shanlin, Peters Ralph S, Podsiadlowski Lars, Vasilikopoulos Alexandros, Zhou Xin, Misof Bernhard, Niehuis Oliver
Center for Molecular Biodiversity Research, Zoological Research Museum Alexander Koenig, Adenauerallee 160, Bonn, 53113, Germany.
Australian National Insect Collection, CSIRO National Research Collections Australia (NRCA), Clunies Ross Street, Canberra, ACT 2601, Australia.
BMC Bioinformatics. 2017 Feb 16;18(1):111. doi: 10.1186/s12859-017-1529-8.
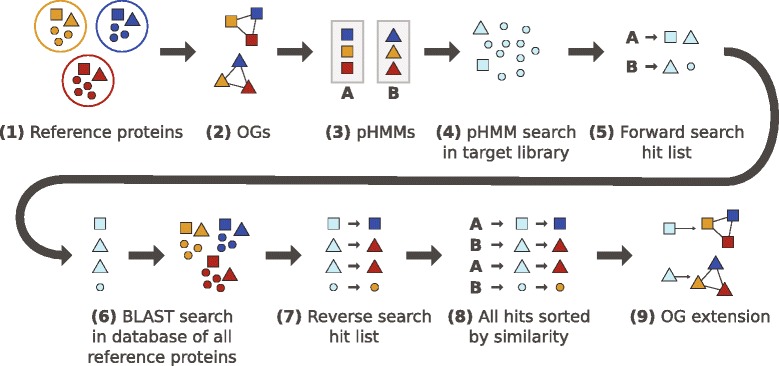
Orthology characterizes genes of different organisms that arose from a single ancestral gene via speciation, in contrast to paralogy, which is assigned to genes that arose via gene duplication. An accurate orthology assignment is a crucial step for comparative genomic studies. Orthologous genes in two organisms can be identified by applying a so-called reciprocal search strategy, given that complete information of the organisms' gene repertoire is available. In many investigations, however, only a fraction of the gene content of the organisms under study is examined (e.g., RNA sequencing). Here, identification of orthologous nucleotide or amino acid sequences can be achieved using a graph-based approach that maps nucleotide sequences to genes of known orthology. Existing implementations of this approach, however, suffer from algorithmic issues that may cause problems in downstream analyses.
We present a new software pipeline, Orthograph, that addresses and solves the above problems and implements useful features for a wide range of comparative genomic and transcriptomic analyses. Orthograph applies a best reciprocal hit search strategy using profile hidden Markov models and maps nucleotide sequences to the globally best matching cluster of orthologous genes, thus enabling researchers to conveniently and reliably delineate orthologs and paralogs from transcriptomic and genomic sequence data. We demonstrate the performance of our approach on de novo-sequenced and assembled transcript libraries of 24 species of apoid wasps (Hymenoptera: Aculeata) as well as on published genomic datasets.
With Orthograph, we implemented a best reciprocal hit approach to reference-based orthology prediction for coding nucleotide sequences such as RNAseq data. Orthograph is flexible, easy to use, open source and freely available at https://mptrsen.github.io/Orthograph . Additionally, we release 24 de novo-sequenced and assembled transcript libraries of apoid wasp species.
直系同源性描述的是不同生物体中通过物种形成从单个祖先基因衍生而来的基因,与之相对的是旁系同源性,旁系同源性指的是通过基因复制产生的基因。准确的直系同源性分配是比较基因组研究的关键步骤。如果有生物体基因库的完整信息,那么可以通过应用所谓的相互搜索策略来识别两种生物体中的直系同源基因。然而,在许多研究中,仅检查了所研究生物体基因内容的一部分(例如RNA测序)。在这里,可以使用基于图形的方法来识别直系同源核苷酸或氨基酸序列,该方法将核苷酸序列映射到已知直系同源性的基因。然而,该方法的现有实现存在算法问题,可能会在下游分析中导致问题。
我们提出了一种新的软件流程Orthograph,它解决了上述问题,并为广泛的比较基因组和转录组分析实现了有用的功能。Orthograph使用轮廓隐马尔可夫模型应用最佳相互比对搜索策略,并将核苷酸序列映射到直系同源基因的全局最佳匹配簇,从而使研究人员能够方便且可靠地从转录组和基因组序列数据中划分出直系同源基因和旁系同源基因。我们在24种apoide黄蜂(膜翅目:针尾部)的从头测序和组装转录本库以及已发表的基因组数据集上展示了我们方法的性能。
通过Orthograph,我们实现了一种基于参考的直系同源性预测的最佳相互比对方法,用于诸如RNAseq数据等编码核苷酸序列。Orthograph灵活、易于使用、开源且可在https://mptrsen.github.io/Orthograph上免费获取。此外,我们发布了24种apoide黄蜂物种的从头测序和组装转录本库。