Human Genome Center, Institute of Medical Science, The University of Tokyo, 4-6-1 Shirokane-dai, Minato-ku, Tokyo 108-8639, Japan.
Bioinformatics. 2009 Oct 15;25(20):2677-84. doi: 10.1093/bioinformatics/btp442. Epub 2009 Jul 20.
Recent improvements in DNA microarray techniques have made a large variety of gene expression data available in public databases. This data can be used to evaluate the strength of gene coexpression by calculating the correlation of expression patterns among different genes between many experiments. However, gene expression levels differ significantly across various tissues in higher organisms, as well as in different cellular location in eukaryotes in different cell state. Thus the usual correlation measure can only evaluate the difference of tissues or cellular localizations, and cannot adequately elucidate the functional relationship from the coexpression of genes.
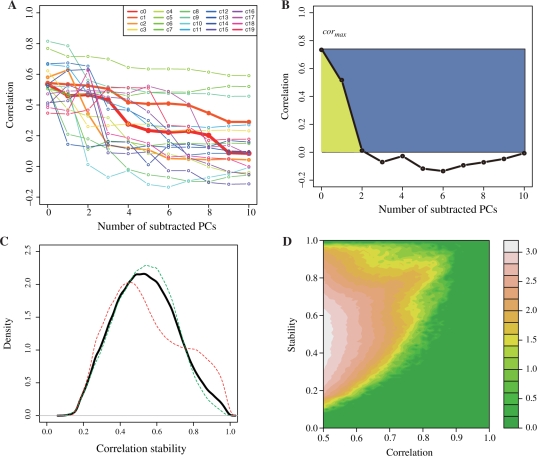
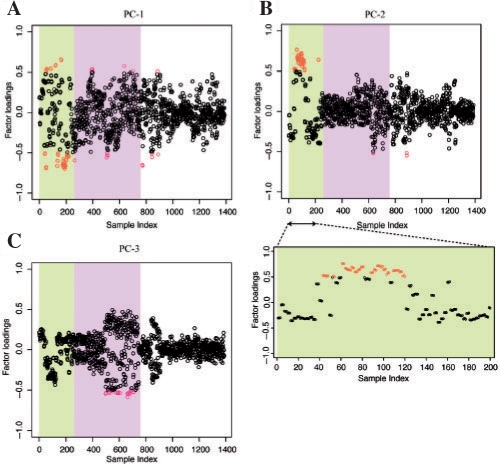
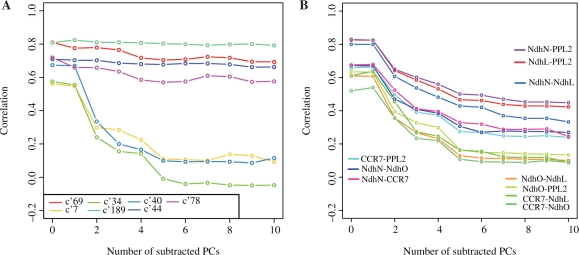
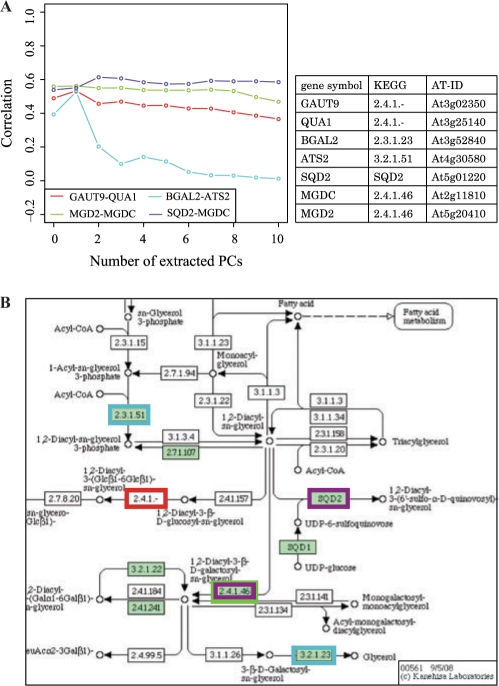
We propose a new measure of coexpression by expanding the generally used correlation into a multidimensional one. We used principal component analyses to identify the major factors of gene expression correlation, and then re-calculate the correlation by subtracting the major components in order to remove biases cased by a few experiments. The repeated subtractions of the major components yielded a set of correlation values for each pair of genes. We observed the correlation changes when the first ten principal components were subtracted step-by-step in large-scale Arabidopsis expression data.
We found two extreme patterns of correlation changes, corresponding to stable and fragile coexpression. Our new indexes provided a good means to determine the functional relationships of the genes, by examining a few examples, and higher performance of Gene Ontology term prediction by using the support vector machine and the multidimensional correlation.
The results are available from the expression detail pages in ATTED-II (http://atted.jp).
最近 DNA 微阵列技术的改进使得大量基因表达数据可在公共数据库中使用。通过计算不同实验中不同基因之间表达模式的相关性,可以利用这些数据来评估基因共表达的强度。然而,在高等生物的各种组织中以及真核生物的不同细胞位置和不同细胞状态中,基因表达水平存在显著差异。因此,通常的相关度量方法只能评估组织或细胞定位的差异,而不能充分阐明基因共表达的功能关系。
我们通过将常用的相关性扩展为多维相关性来提出一种新的共表达度量方法。我们使用主成分分析来识别基因表达相关性的主要因素,然后通过减去主要成分来重新计算相关性,以消除少数实验引起的偏差。重复减去主要成分可得到每对基因的一组相关值。我们在大规模拟南芥表达数据中观察到当逐步减去前十个主成分时相关性的变化。
我们发现了两种极端的相关性变化模式,分别对应于稳定和脆弱的共表达。我们的新指标通过检查几个例子,为确定基因的功能关系提供了一种很好的方法,并且通过使用支持向量机和多维相关性,提高了基因本体论术语预测的性能。
结果可从 ATTED-II 的表达详细信息页面获得(http://atted.jp)。