Göker Markus, García-Blázquez Gema, Voglmayr Hermann, Tellería M Teresa, Martín María P
Organismic Botany, Eberhard Karls University of Tübingen, Tübingen, Germany.
PLoS One. 2009 Jul 29;4(7):e6319. doi: 10.1371/journal.pone.0006319.
Inappropriate taxon definitions may have severe consequences in many areas. For instance, biologically sensible species delimitation of plant pathogens is crucial for measures such as plant protection or biological control and for comparative studies involving model organisms. However, delimiting species is challenging in the case of organisms for which often only molecular data are available, such as prokaryotes, fungi, and many unicellular eukaryotes. Even in the case of organisms with well-established morphological characteristics, molecular taxonomy is often necessary to emend current taxonomic concepts and to analyze DNA sequences directly sampled from the environment. Typically, for this purpose clustering approaches to delineate molecular operational taxonomic units have been applied using arbitrary choices regarding the distance threshold values, and the clustering algorithms.
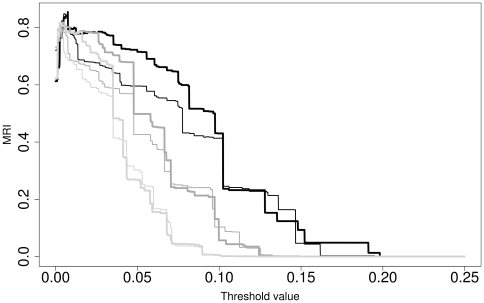
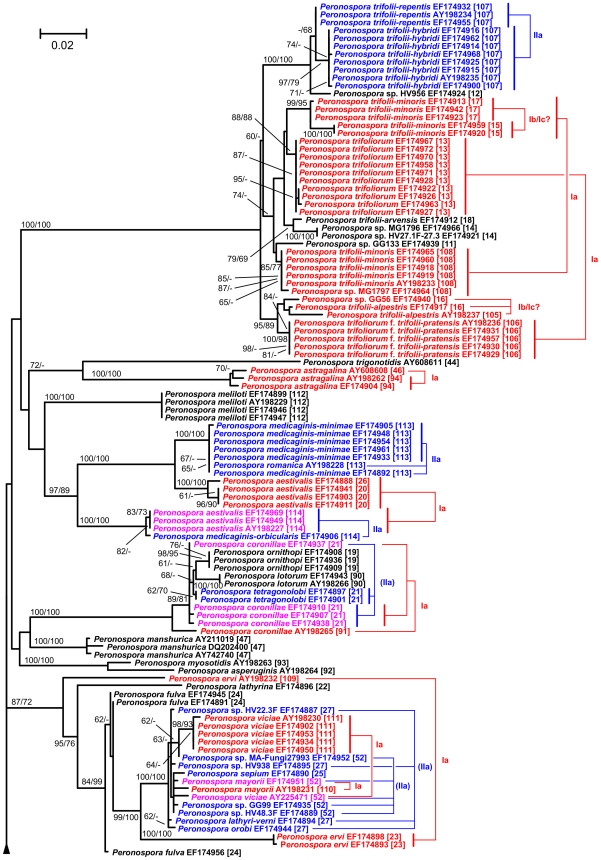
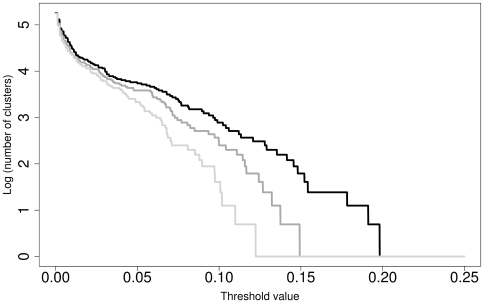
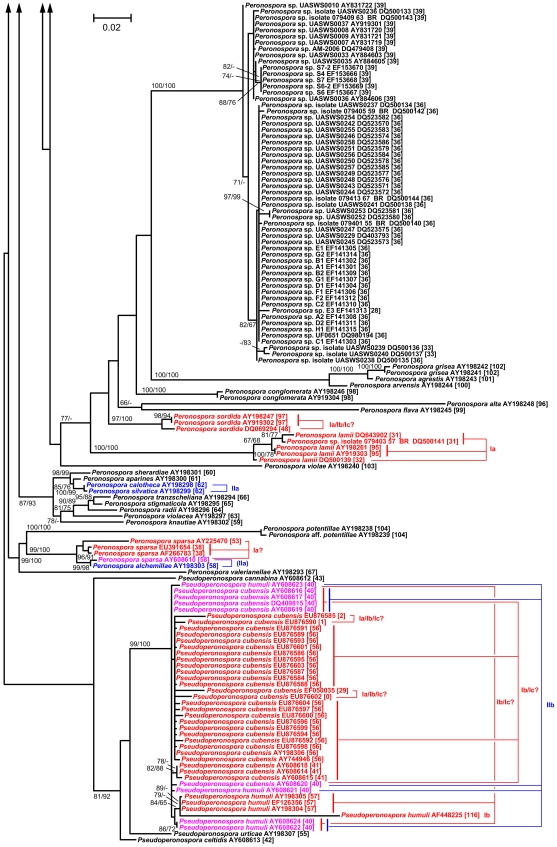
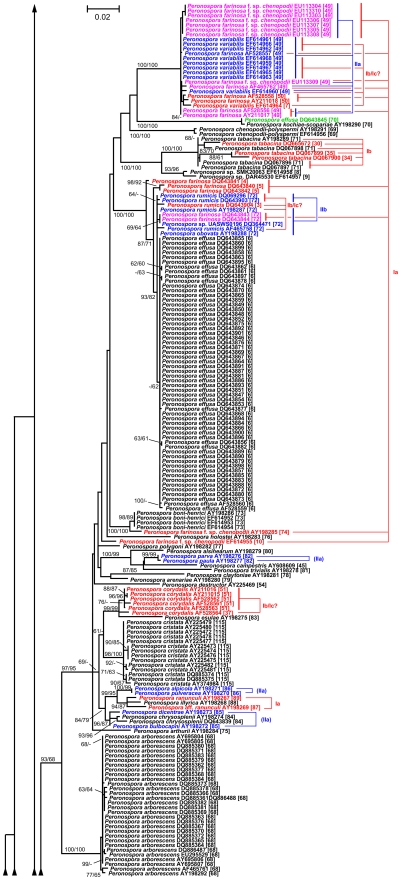
Here, we report on a clustering optimization method to establish a molecular taxonomy of Peronospora based on ITS nrDNA sequences. Peronospora is the largest genus within the downy mildews, which are obligate parasites of higher plants, and includes various economically important pathogens. The method determines the distance function and clustering setting that result in an optimal agreement with selected reference data. Optimization was based on both taxonomy-based and host-based reference information, yielding the same outcome. Resampling and permutation methods indicate that the method is robust regarding taxon sampling and errors in the reference data. Tests with newly obtained ITS sequences demonstrate the use of the re-classified dataset in molecular identification of downy mildews.
A corrected taxonomy is provided for all Peronospora ITS sequences contained in public databases. Clustering optimization appears to be broadly applicable in automated, sequence-based taxonomy. The method connects traditional and modern taxonomic disciplines by specifically addressing the issue of how to optimally account for both traditional species concepts and genetic divergence.
不恰当的分类单元定义可能在许多领域产生严重后果。例如,对植物病原体进行生物学上合理的物种界定对于植物保护或生物防治等措施以及涉及模式生物的比较研究至关重要。然而,对于通常仅能获得分子数据的生物,如原核生物、真菌和许多单细胞真核生物,界定物种具有挑战性。即使对于具有明确形态特征的生物,分子分类学通常也是必要的,以修正当前的分类学概念并直接分析从环境中采样的DNA序列。通常,为此目的,已应用聚类方法来划定分子操作分类单元,使用了关于距离阈值和聚类算法的任意选择。
在此,我们报告一种基于ITS nrDNA序列建立霜霉属分子分类学的聚类优化方法。霜霉属是霜霉目中最大的属,是高等植物的专性寄生菌,包括各种具有重要经济意义的病原体。该方法确定导致与选定参考数据最佳匹配的距离函数和聚类设置。优化基于基于分类学和基于宿主的参考信息,产生相同的结果。重采样和置换方法表明该方法在分类单元采样和参考数据误差方面具有稳健性。对新获得的ITS序列进行的测试证明了重新分类数据集在霜霉病分子鉴定中的应用。
为公共数据库中包含的所有霜霉属ITS序列提供了修正的分类学。聚类优化似乎广泛适用于基于序列的自动化分类学。该方法通过专门解决如何最佳兼顾传统物种概念和遗传差异的问题,将传统和现代分类学科联系起来。