Wong Jason W H, Schwahn Alexander B, Downard Kevin M
UNSW Cancer Research Centre, University of New South Wales, Sydney, NSW 2052, Australia.
BMC Bioinformatics. 2009 Aug 10;10:244. doi: 10.1186/1471-2105-10-244.
Concurrent peptide fragmentation (i.e. shotgun CID, parallel CID or MSE) has emerged as an alternative to data-dependent acquisition in generating peptide fragmentation data in LC-MS/MS proteomics experiments. Concurrent peptide fragmentation data acquisition has been shown to be advantageous over data-dependent acquisition by providing greater detection dynamic range and providing more accurate quantitative information. Nevertheless, concurrent peptide fragmentation data acquisition remains to be widely adopted due to the lack of published algorithms designed specifically to process or interpret such data acquired on any mass spectrometer.
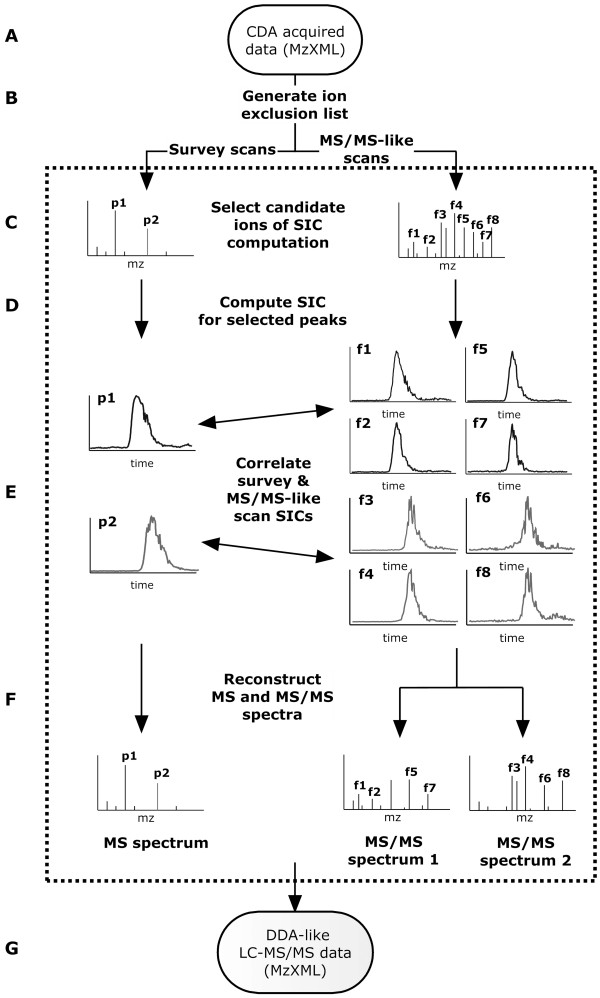
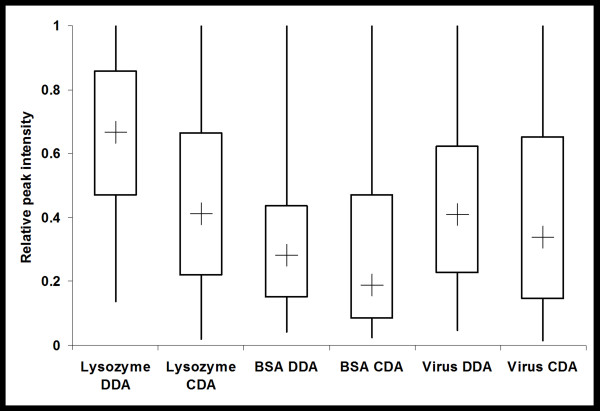
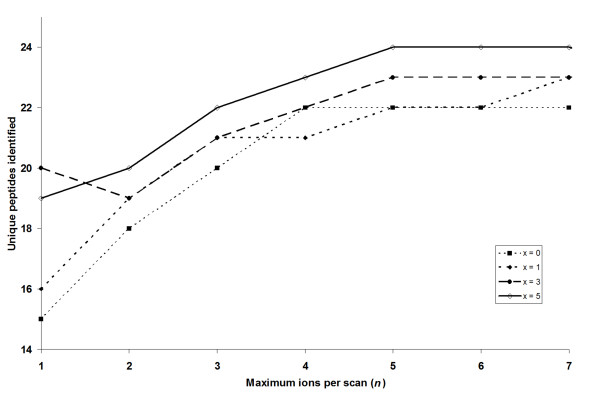
An algorithm called Elution Time Ion Sequencing (ETISEQ), has been developed to enable automated conversion of concurrent peptide fragmentation data acquisition data to LC-MS/MS data. ETISEQ generates MS/MS-like spectra based on the correlation of precursor and product ion elution profiles. The performance of ETISEQ is demonstrated using concurrent peptide fragmentation data from tryptic digests of standard proteins and whole influenza virus. It is shown that the number of unique peptides identified from the digests is broadly comparable between ETISEQ processed concurrent peptide fragmentation data and the data-dependent acquired LC-MS/MS data.
The ETISEQ algorithm has been designed for easy integration with existing MS/MS analysis platforms. It is anticipated that it will popularize concurrent peptide fragmentation data acquisition in proteomics laboratories.
在液相色谱-串联质谱(LC-MS/MS)蛋白质组学实验中,并行肽段碎裂(即鸟枪法碰撞诱导解离、平行碰撞诱导解离或质谱选择性激发)已成为一种替代数据依赖型采集来生成肽段碎裂数据的方法。并行肽段碎裂数据采集已被证明比数据依赖型采集更具优势,它能提供更大的检测动态范围并提供更准确的定量信息。然而,由于缺乏专门设计用于处理或解释在任何质谱仪上采集的此类数据的已发表算法,并行肽段碎裂数据采集仍未得到广泛应用。
一种名为洗脱时间离子测序(ETISEQ)的算法已被开发出来,用于将并行肽段碎裂数据采集数据自动转换为LC-MS/MS数据。ETISEQ基于前体离子和产物离子洗脱图谱的相关性生成类似串联质谱的谱图。使用来自标准蛋白质和全流感病毒胰蛋白酶消化物的并行肽段碎裂数据展示了ETISEQ的性能。结果表明,从消化物中鉴定出的独特肽段数量在ETISEQ处理的并行肽段碎裂数据和数据依赖型采集的LC-MS/MS数据之间大致相当。
ETISEQ算法设计为易于与现有的串联质谱分析平台集成。预计它将在蛋白质组学实验室中推广并行肽段碎裂数据采集。