MTi, RPBS, INSERM UMR-S973, Université Paris Diderot-Paris 7, Paris, France.
BMC Bioinformatics. 2009 Aug 11;10:245. doi: 10.1186/1471-2105-10-245.
Virtual screening methods are now well established as effective to identify hit and lead candidates and are fully integrated in most drug discovery programs. Ligand-based approaches make use of physico-chemical, structural and energetics properties of known active compounds to search large chemical libraries for related and novel chemotypes. While 2D-similarity search tools are known to be fast and efficient, the use of 3D-similarity search methods can be very valuable to many research projects as integration of "3D knowledge" can facilitate the identification of not only related molecules but also of chemicals possessing distant scaffolds as compared to the query and therefore be more inclined to scaffolds hopping. To date, very few methods performing this task are easily available to the scientific community.
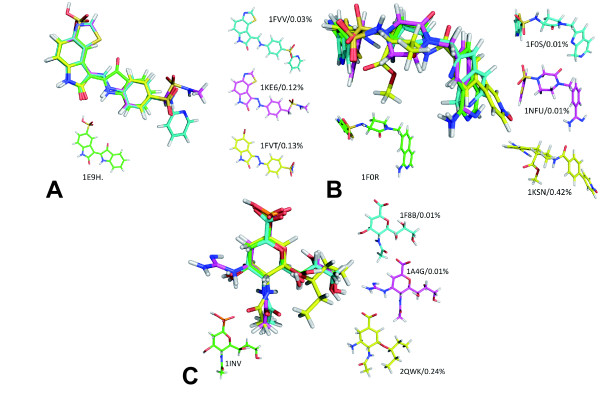
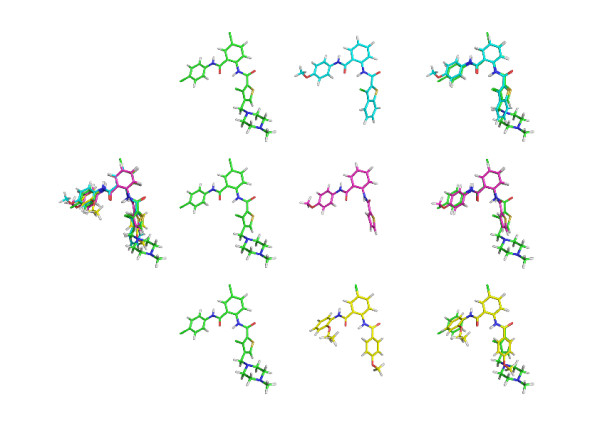
We introduce a new approach (LigCSRre) to the 3D ligand similarity search of drug candidates. It combines a 3D maximum common substructure search algorithm independent on atom order with a tunable description of atomic compatibilities to prune the search and increase its physico-chemical relevance. We show, on 47 experimentally validated active compounds across five protein targets having different specificities, that for single compound search, the approach is able to recover on average 52% of the co-actives in the top 1% of the ranked list which is better than gold standards of the field. Moreover, the combination of several runs on a single protein target using different query active compounds shows a remarkable improvement in enrichment. Such Results demonstrate LigCSRre as a valuable tool for ligand-based screening.
LigCSRre constitutes a new efficient and generic approach to the 3D similarity screening of small compounds, whose flexible design opens the door to many enhancements. The program is freely available to the academics for non-profit research at: http://bioserv.rpbs.univ-paris-diderot.fr/LigCSRre.html.
虚拟筛选方法现已被广泛应用于识别命中化合物和先导化合物,并且已在大多数药物发现项目中得到充分整合。配体方法利用已知活性化合物的物理化学、结构和能量性质,在大型化学库中搜索相关和新颖的化学型。虽然二维相似性搜索工具速度快、效率高,但 3D 相似性搜索方法的使用对许多研究项目非常有价值,因为“3D 知识”的整合不仅可以帮助识别相关分子,还可以识别与查询相比具有较远骨架的化学物质,从而更倾向于骨架跳跃。迄今为止,科学界可轻松获得的执行此任务的方法非常少。
我们引入了一种新的方法(LigCSRre),用于药物候选物的 3D 配体相似性搜索。它结合了一个不依赖于原子顺序的 3D 最大公共子结构搜索算法和一个可调节的原子兼容性描述,以修剪搜索并提高其物理化学相关性。我们在五个具有不同特异性的蛋白质靶标上的 47 个经过实验验证的活性化合物上进行了研究,结果表明,对于单个化合物搜索,该方法能够在排名前 1%的列表中平均恢复 52%的共同活性化合物,优于该领域的黄金标准。此外,在单个蛋白质靶标上使用不同查询活性化合物进行多次运行的组合显示出显著的富集提高。这些结果表明,LigCSRre 是一种用于配体筛选的有价值的工具。
LigCSRre 是一种新的有效和通用的小分子 3D 相似性筛选方法,其灵活的设计为许多增强功能打开了大门。该程序可在以下网址免费供学术界用于非营利性研究:http://bioserv.rpbs.univ-paris-diderot.fr/LigCSRre.html。