Gal Jozsef, Ström Anna-Lena, Kwinter David M, Kilty Renée, Zhang Jiayu, Shi Ping, Fu Weisi, Wooten Marie W, Zhu Haining
Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, Kentucky 40536, USA.
J Neurochem. 2009 Nov;111(4):1062-73. doi: 10.1111/j.1471-4159.2009.06388.x. Epub 2009 Sep 18.
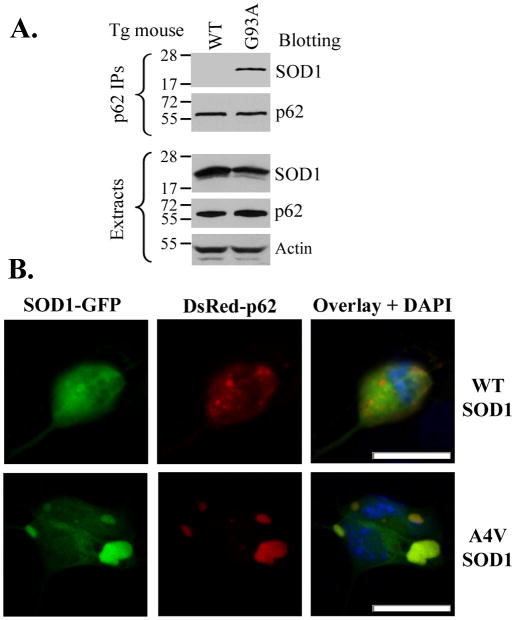
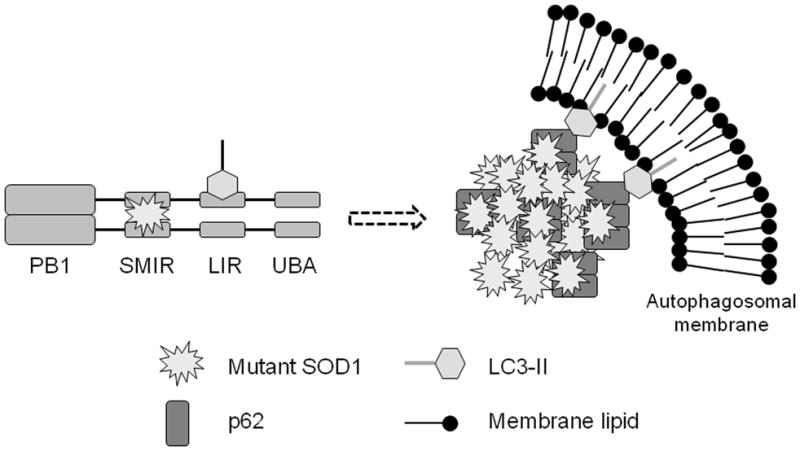
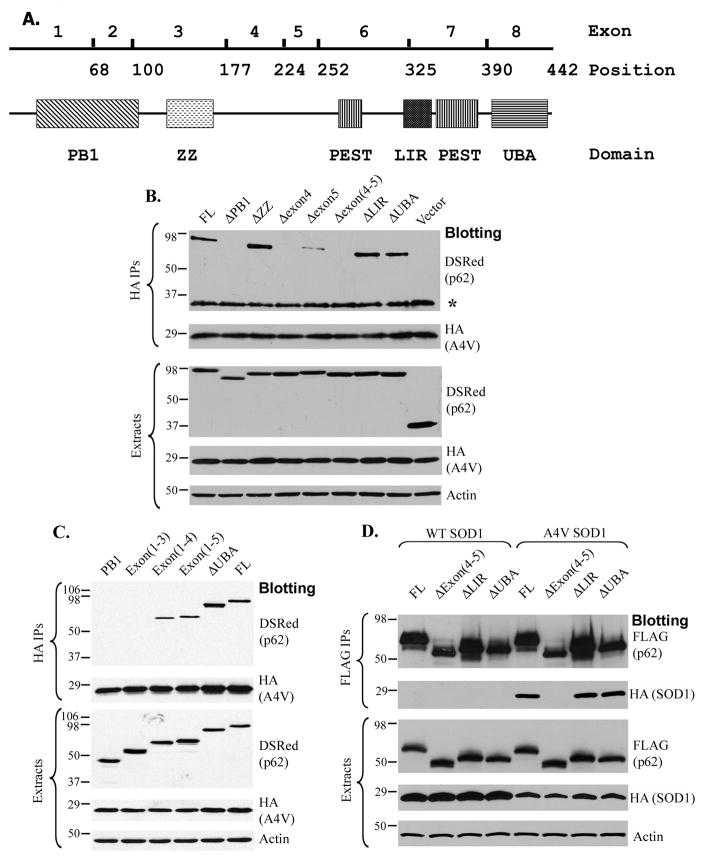
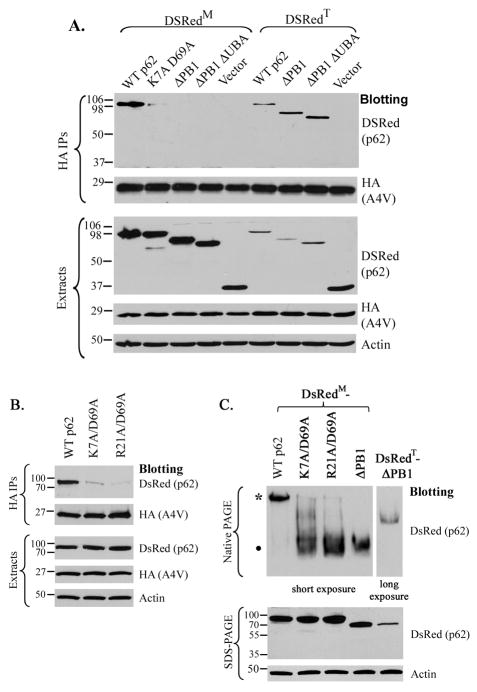
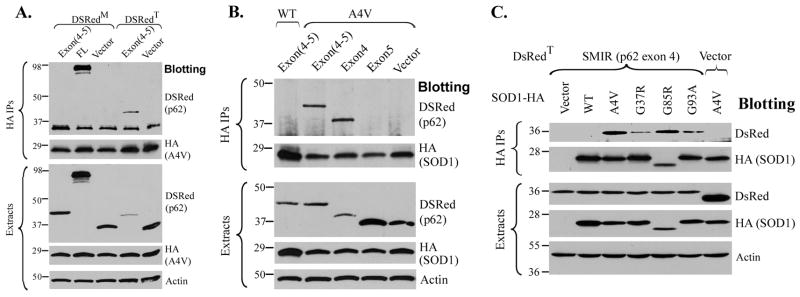
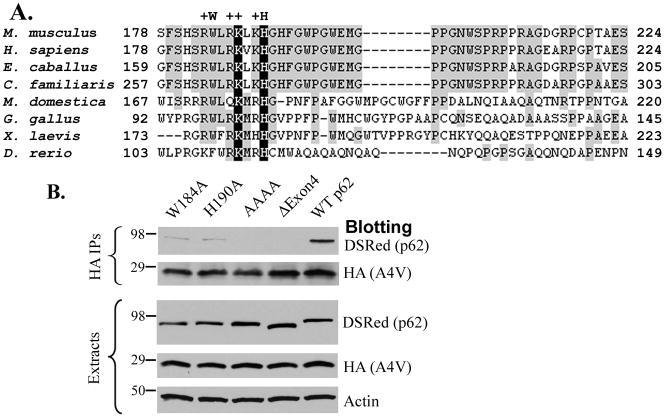
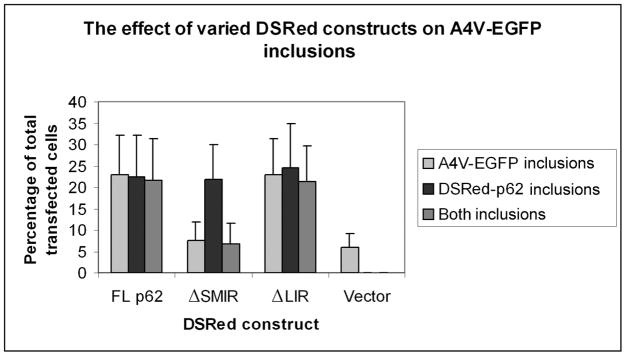
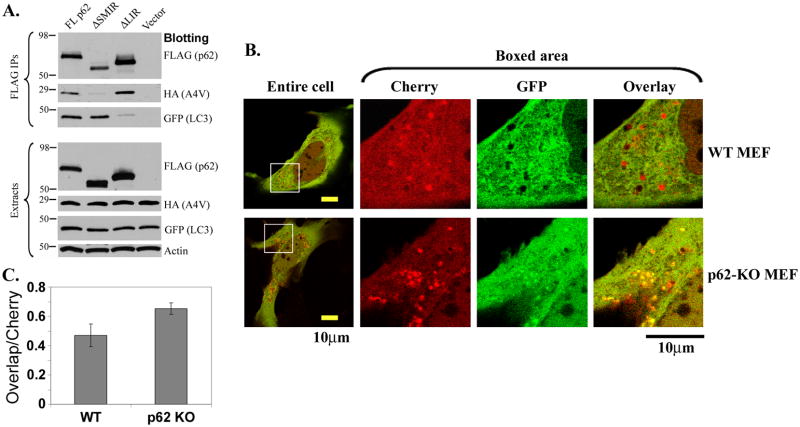
The p62/sequestosome 1 protein has been identified as a component of pathological protein inclusions in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). P62 has also been implicated in autophagy, a process of mass degradation of intracellular proteins and organelles. Autophagy is a critical pathway for degrading misfolded and/or damaged proteins, including the copper-zinc superoxide dismutase (SOD1) mutants linked to familial ALS. We previously reported that p62 interacted with ALS mutants of SOD1 and that the ubiquitin-association domain of p62 was dispensable for the interaction. In this study, we identified two distinct regions of p62 that were essential to its binding to mutant SOD1: the N-terminal Phox and Bem1 (PB1) domain (residues 1-104) and a separate internal region (residues 178-224) termed here as SOD1 mutant interaction region (SMIR). The PB1 domain is required for appropriate oligomeric status of p62 and the SMIR is the actual region interacting with mutant SOD1. Within the SMIR, the conserved W184, H190 and positively charged R183, R186, K187, and K189 residues are critical to the p62-mutant SOD1 interaction as substitution of these residues with alanine resulted in significantly abolished binding. In addition, SMIR and the p62 sequence responsible for the interaction with LC3, a protein essential for autophagy activation, are independent of each other. In cells lacking p62, the existence of mutant SOD1 in acidic autolysosomes decreased, suggesting that p62 can function as an adaptor between mutant SOD1 and the autophagy machinery. This study provides a novel molecular mechanism by which mutant SOD1 can be recognized by p62 in an ubiquitin-independent fashion and targeted for the autophagy-lysosome degradation pathway.
p62/聚集体包含蛋白1已被确定为神经退行性疾病(包括肌萎缩侧索硬化症,即ALS)中病理性蛋白聚集体的一个组成部分。P62也与自噬有关,自噬是细胞内蛋白质和细胞器大量降解的过程。自噬是降解错误折叠和/或受损蛋白质(包括与家族性ALS相关的铜锌超氧化物歧化酶(SOD1)突变体)的关键途径。我们之前报道过p62与SOD1的ALS突变体相互作用,并且p62的泛素结合结构域对于这种相互作用是可有可无的。在本研究中,我们确定了p62与突变型SOD1结合所必需的两个不同区域:N端的Phox和Bem1(PB1)结构域(第1 - 104位氨基酸)以及一个单独的内部区域(第178 - 224位氨基酸),在此称为SOD1突变体相互作用区域(SMIR)。PB1结构域对于p62适当的寡聚状态是必需的,而SMIR是与突变型SOD1实际相互作用的区域。在SMIR内,保守的W184、H190以及带正电荷的R183、R186、K187和K189氨基酸残基对于p62 - 突变型SOD1相互作用至关重要,因为用丙氨酸替代这些残基会导致结合显著消失。此外,SMIR和负责与LC3(自噬激活所必需的一种蛋白质)相互作用的p62序列彼此独立。在缺乏p62的细胞中,酸性自噬溶酶体中突变型SOD1的存在减少,这表明p62可以作为突变型SOD1与自噬机制之间的衔接蛋白发挥作用。这项研究提供了一种新的分子机制,通过该机制突变型SOD1能够以不依赖泛素的方式被p62识别,并被靶向自噬 - 溶酶体降解途径。