Clinical Epidemiology, Biostatistics & Bioinformatics, Academic Medical Center, University of Amsterdam, PO Box 22700, 1100 DE Amsterdam, the Netherlands.
BMC Bioinformatics. 2009 Sep 28;10:315. doi: 10.1186/1471-2105-10-315.
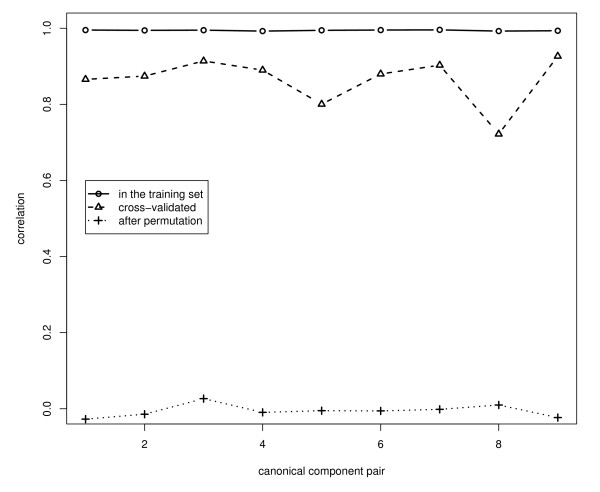
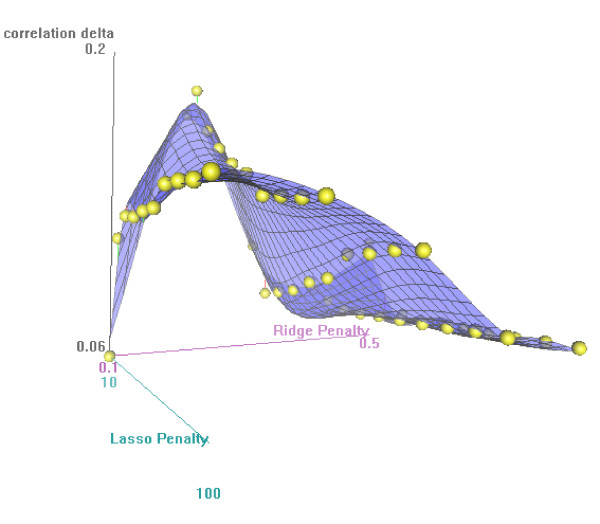
We generalized penalized canonical correlation analysis for analyzing microarray gene-expression measurements for checking completeness of known metabolic pathways and identifying candidate genes for incorporation in the pathway. We used Wold's method for calculation of the canonical variates, and we applied ridge penalization to the regression of pathway genes on canonical variates of the non-pathway genes, and the elastic net to the regression of non-pathway genes on the canonical variates of the pathway genes.
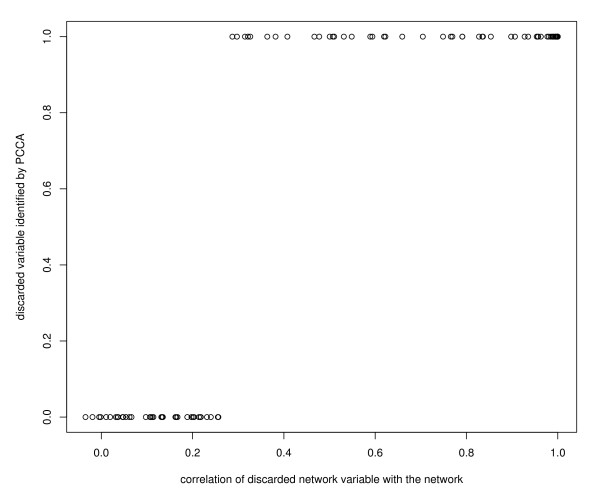
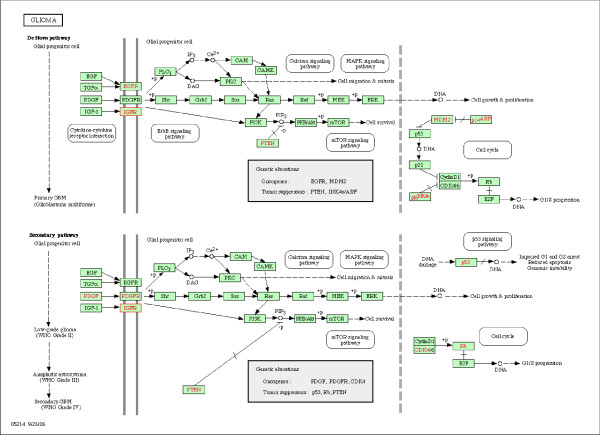
We performed a small simulation to illustrate the model's capability to identify new candidate genes to incorporate in the pathway: in our simulations it appeared that a gene was correctly identified if the correlation with the pathway genes was 0.3 or more. We applied the methods to a gene-expression microarray data set of 12, 209 genes measured in 45 patients with glioblastoma, and we considered genes to incorporate in the glioma-pathway: we identified more than 25 genes that correlated > 0.9 with canonical variates of the pathway genes.
We concluded that penalized canonical correlation analysis is a powerful tool to identify candidate genes in pathway analysis.
我们推广了惩罚典型相关分析,以分析微阵列基因表达测量,以检查已知代谢途径的完整性,并确定候选基因纳入途径。我们使用 Wold 方法计算典型变量,对途径基因与非途径基因的典型变量进行岭惩罚回归,对非途径基因与途径基因的典型变量进行弹性网络回归。
我们进行了一项小型模拟研究,以说明该模型识别新的候选基因以纳入途径的能力:在我们的模拟中,如果与途径基因的相关性为 0.3 或更高,则可以正确识别基因。我们将该方法应用于 45 名胶质母细胞瘤患者的 12209 个基因的基因表达微阵列数据集,并考虑将基因纳入神经胶质瘤途径:我们确定了 25 个以上与途径基因的典型变量相关系数> 0.9 的基因。
我们得出结论,惩罚典型相关分析是一种在途径分析中识别候选基因的强大工具。