Swiss Institute for Experimental Cancer Research (ISREC), Swiss Federal Institute of Technology (EPFL), School of Life Sciences, EPFL SV ISREC, Lausanne, Switzerland.
PLoS One. 2009 Oct 23;4(10):e7431. doi: 10.1371/journal.pone.0007431.
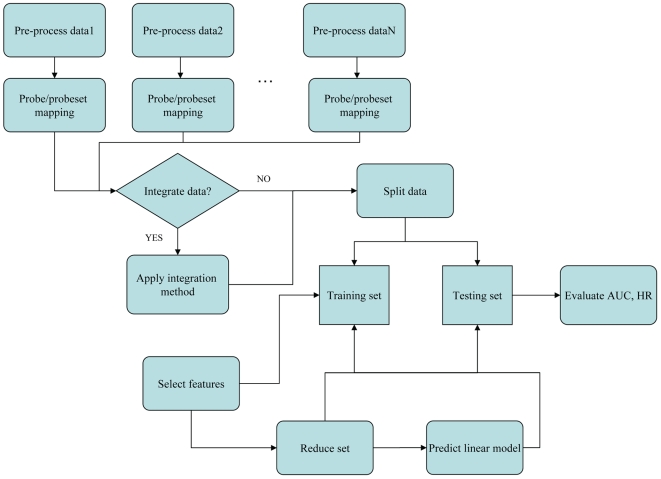
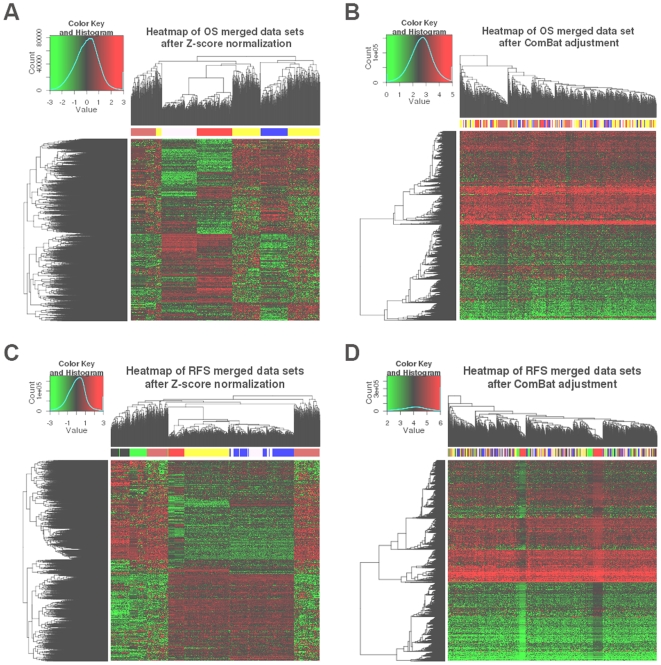
High-throughput gene expression profiling technologies generating a wealth of data, are increasingly used for characterization of tumor biopsies for clinical trials. By applying machine learning algorithms to such clinically documented data sets, one hopes to improve tumor diagnosis, prognosis, as well as prediction of treatment response. However, the limited number of patients enrolled in a single trial study limits the power of machine learning approaches due to over-fitting. One could partially overcome this limitation by merging data from different studies. Nevertheless, such data sets differ from each other with regard to technical biases, patient selection criteria and follow-up treatment. It is therefore not clear at all whether the advantage of increased sample size outweighs the disadvantage of higher heterogeneity of merged data sets. Here, we present a systematic study to answer this question specifically for breast cancer data sets. We use survival prediction based on Cox regression as an assay to measure the added value of merged data sets.
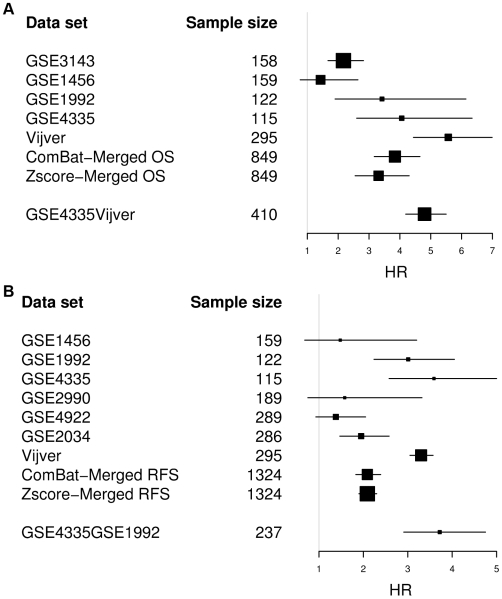
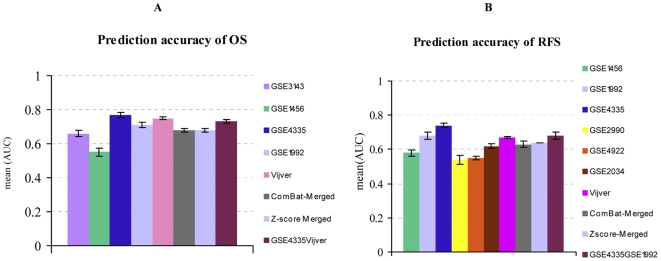
Using time-dependent Receiver Operating Characteristic-Area Under the Curve (ROC-AUC) and hazard ratio as performance measures, we see in overall no significant improvement or deterioration of survival prediction with merged data sets as compared to individual data sets. This apparently was due to the fact that a few genes with strong prognostic power were not available on all microarray platforms and thus were not retained in the merged data sets. Surprisingly, we found that the overall best performance was achieved with a single-gene predictor consisting of CYB5D1.
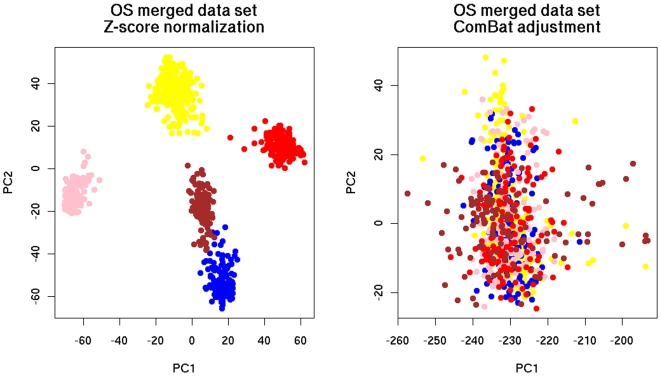
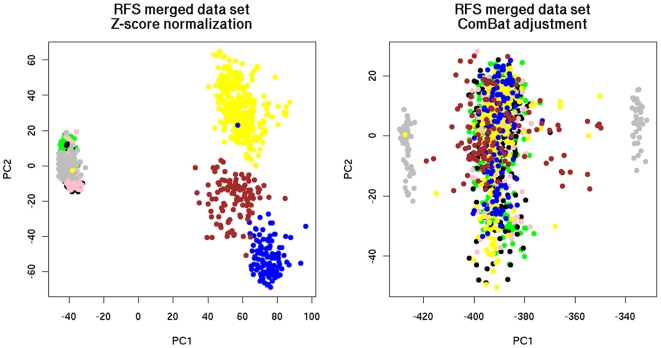
Merging did not deteriorate performance on average despite (a) The diversity of microarray platforms used. (b) The heterogeneity of patients cohorts. (c) The heterogeneity of breast cancer disease. (d) Substantial variation of time to death or relapse. (e) The reduced number of genes in the merged data sets. Predictors derived from the merged data sets were more robust, consistent and reproducible across microarray platforms. Moreover, merging data sets from different studies helps to better understand the biases of individual studies and can lead to the identification of strong survival factors like CYB5D1 expression.
高通量基因表达谱技术产生了大量的数据,越来越多地用于临床试验中的肿瘤活检的特征描述。通过将机器学习算法应用于这些有临床记录的数据集中,人们希望能够改善肿瘤的诊断、预后以及治疗反应的预测。然而,由于过度拟合,单个试验研究中纳入的患者数量有限限制了机器学习方法的能力。通过合并来自不同研究的数据,可以部分克服这一限制。然而,这些数据集在技术偏差、患者选择标准和随访治疗方面存在差异。因此,增加样本量的优势是否超过合并数据集异质性增加的劣势还远不清楚。在这里,我们专门针对乳腺癌数据集进行了一项系统研究来回答这个问题。我们使用基于 Cox 回归的生存预测作为衡量合并数据集增加价值的检测方法。
使用时间依赖性接收器工作特性曲线下面积(ROC-AUC)和危险比作为性能指标,我们发现与单个数据集相比,合并数据集的生存预测没有明显的改善或恶化。这显然是因为一些具有很强预后能力的基因在所有微阵列平台上都不可用,因此在合并的数据集中没有保留。令人惊讶的是,我们发现由 CYB5D1 组成的单个基因预测器的总体性能最佳。
尽管存在以下因素,合并数据集平均上没有降低性能:(a)使用的微阵列平台的多样性;(b)患者队列的异质性;(c)乳腺癌疾病的异质性;(d)死亡或复发的时间变化很大;(e)合并数据集中的基因数量减少。从合并数据集中得出的预测器在微阵列平台之间更稳健、一致且可重复。此外,合并来自不同研究的数据有助于更好地理解单个研究的偏差,并可以识别像 CYB5D1 表达这样的强生存因素。