School of Biosciences, University of Exeter, Exeter EX4 5DE, UK.
BMC Bioinformatics. 2009 Oct 29;10:361. doi: 10.1186/1471-2105-10-361.
Tyrosine sulfation is one of the most important posttranslational modifications. Due to its relevance to various disease developments, tyrosine sulfation has become the target for drug design. In order to facilitate efficient drug design, accurate prediction of sulfotyrosine sites is desirable. A predictor published seven years ago has been very successful with claimed prediction accuracy of 98%. However, it has a particularly low sensitivity when predicting sulfotyrosine sites in some newly sequenced proteins.
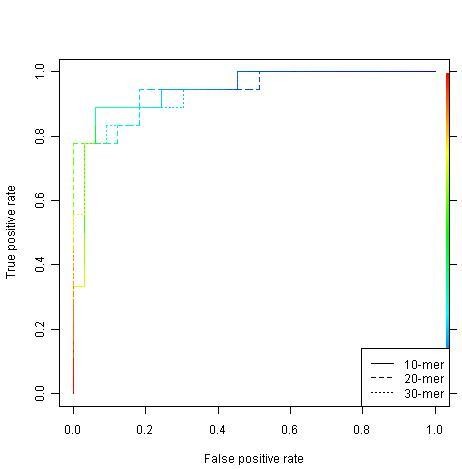
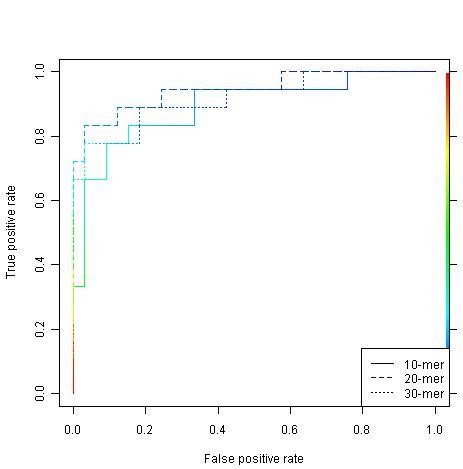
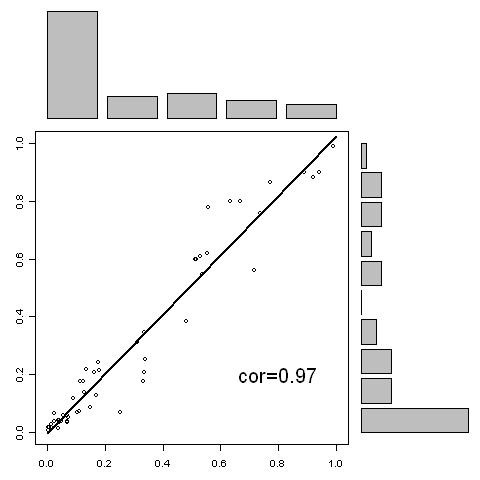
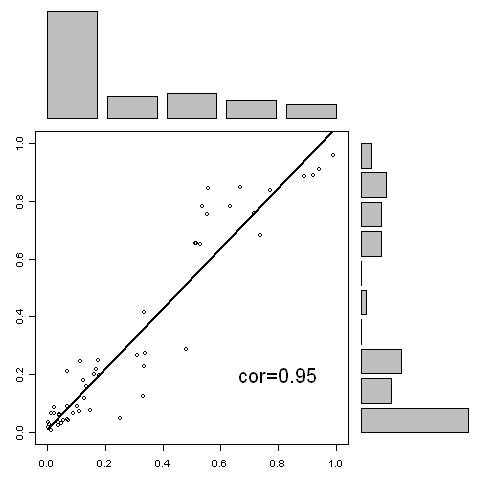
A new approach has been developed for predicting sulfotyrosine sites using the random forest algorithm after a careful evaluation of seven machine learning algorithms. Peptides are formed by consecutive residues symmetrically flanking tyrosine sites. They are then encoded using an amino acid hydrophobicity scale. This new approach has increased the sensitivity by 22%, the specificity by 3%, and the total prediction accuracy by 10% compared with the previous predictor using the same blind data. Meanwhile, both negative and positive predictive powers have been increased by 9%. In addition, the random forest model has an excellent feature for ranking the residues flanking tyrosine sites, hence providing more information for further investigating the tyrosine sulfation mechanism. A web tool has been implemented at http://ecsb.ex.ac.uk/sulfotyrosine for public use.
The random forest algorithm is able to deliver a better model compared with the Hidden Markov Model, the support vector machine, artificial neural networks, and others for predicting sulfotyrosine sites. The success shows that the random forest algorithm together with an amino acid hydrophobicity scale encoding can be a good candidate for peptide classification.
酪氨酸硫酸化是最重要的翻译后修饰之一。由于其与各种疾病发展的相关性,酪氨酸硫酸化已成为药物设计的目标。为了促进高效的药物设计,准确预测硫酸酪氨酸位点是理想的。七年前发表的一个预测器在声称的预测精度为 98%方面非常成功。然而,在预测一些新测序蛋白质中的硫酸酪氨酸位点时,它的灵敏度特别低。
在仔细评估了七种机器学习算法之后,我们使用随机森林算法开发了一种新的预测硫酸酪氨酸位点的方法。肽由酪氨酸位点两侧连续的残基形成。然后,它们使用氨基酸疏水性尺度进行编码。与使用相同盲数据的先前预测器相比,这种新方法将灵敏度提高了 22%,特异性提高了 3%,总预测精度提高了 10%。同时,阴性和阳性预测值都提高了 9%。此外,随机森林模型具有很好的功能,可以对酪氨酸位点周围的残基进行排序,从而为进一步研究酪氨酸硫酸化机制提供更多信息。一个网络工具已在 http://ecsb.ex.ac.uk/sulfotyrosine 上实现,供公众使用。
与隐马尔可夫模型、支持向量机、人工神经网络等相比,随机森林算法能够为预测硫酸酪氨酸位点提供更好的模型。成功表明,随机森林算法结合氨基酸疏水性尺度编码可以成为肽分类的一个很好的候选者。