Department of Bioscience and Bioinformatics, Kyushu Institute of Technology, 680-4 Kawazu, Iizuka, Fukuoka 820-8502, Japan.
WHO Collaborating Centre on eHealth, UNSW Digital Health, School of Public Health and Community Medicine, Faculty of Medicine, UNSW Sydney, Sydney, NSW 2052, Australia.
Int J Mol Sci. 2021 Mar 8;22(5):2704. doi: 10.3390/ijms22052704.
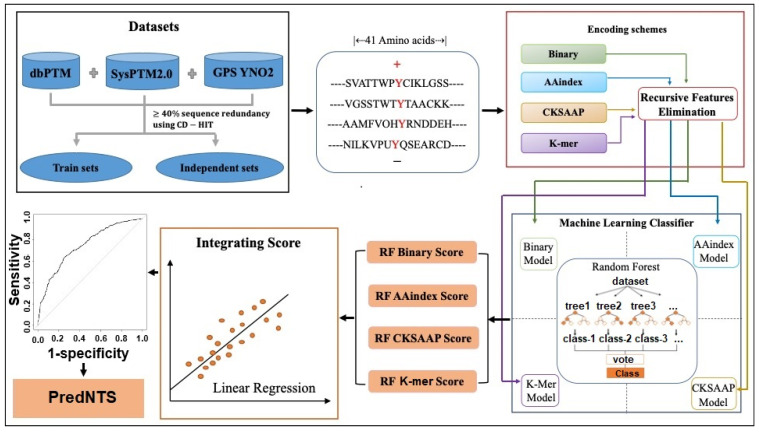
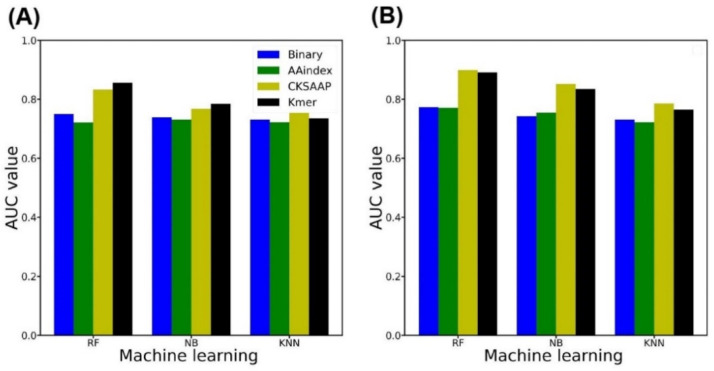
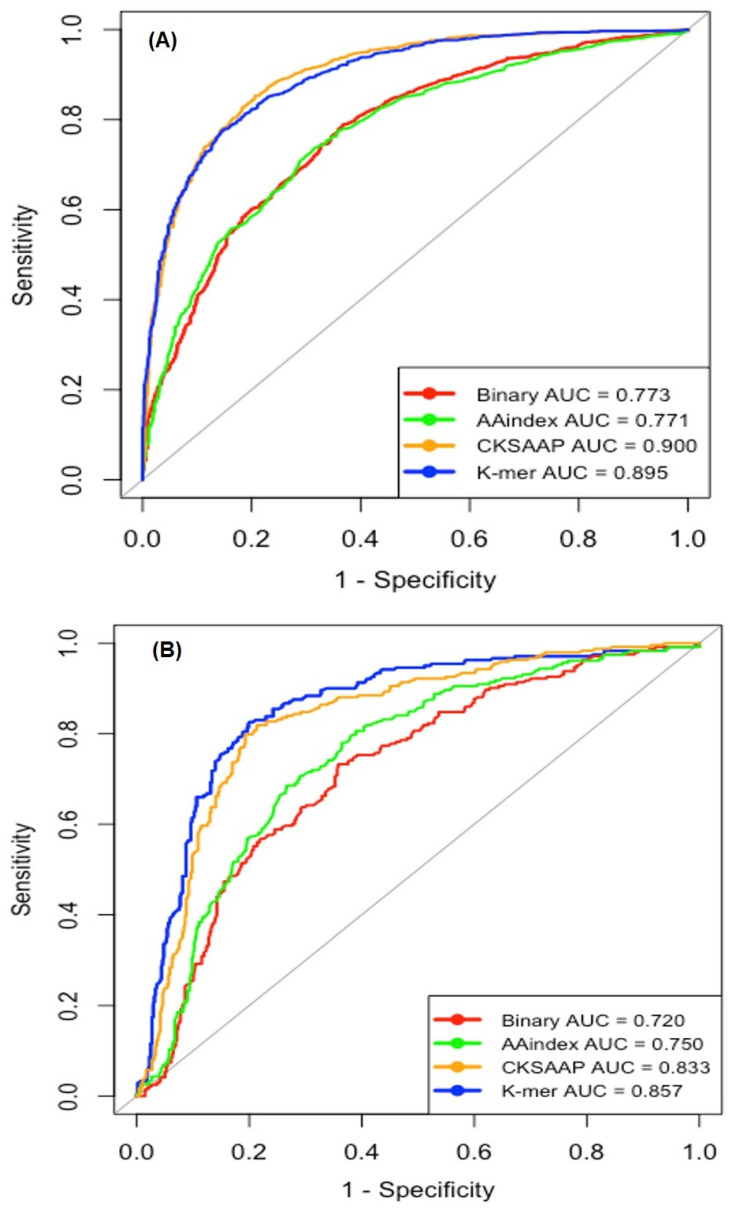
Nitrotyrosine, which is generated by numerous reactive nitrogen species, is a type of protein post-translational modification. Identification of site-specific nitration modification on tyrosine is a prerequisite to understanding the molecular function of nitrated proteins. Thanks to the progress of machine learning, computational prediction can play a vital role before the biological experimentation. Herein, we developed a computational predictor PredNTS by integrating multiple sequence features including K-mer, composition of k-spaced amino acid pairs (CKSAAP), AAindex, and binary encoding schemes. The important features were selected by the recursive feature elimination approach using a random forest classifier. Finally, we linearly combined the successive random forest (RF) probability scores generated by the different, single encoding-employing RF models. The resultant PredNTS predictor achieved an area under a curve (AUC) of 0.910 using five-fold cross validation. It outperformed the existing predictors on a comprehensive and independent dataset. Furthermore, we investigated several machine learning algorithms to demonstrate the superiority of the employed RF algorithm. The PredNTS is a useful computational resource for the prediction of nitrotyrosine sites. The web-application with the curated datasets of the PredNTS is publicly available.
硝酪氨酸是由多种活性氮物种生成的一种蛋白质翻译后修饰类型。鉴定酪氨酸的特异性硝化修饰位点是理解硝化蛋白分子功能的前提。得益于机器学习的进展,计算预测可以在生物学实验之前发挥至关重要的作用。在此,我们通过整合多种序列特征,包括 K -mer、k 间隔氨基酸对组成(CKSAAP)、AAindex 和二进制编码方案,开发了一种名为 PredNTS 的计算预测器。通过使用随机森林分类器的递归特征消除方法选择重要特征。最后,我们线性组合了由不同的、单一编码的随机森林模型生成的连续随机森林(RF)概率得分。该预测器在五重交叉验证中实现了 0.910 的曲线下面积(AUC)。它在综合和独立数据集上优于现有的预测器。此外,我们研究了几种机器学习算法,以证明所采用的 RF 算法的优越性。PredNTS 是预测硝酪氨酸位点的有用计算资源。带有 PredNTS 经过整理的数据集的网络应用程序可供公开使用。