Department of Computer Science, University of California-Davis, Davis, CA 95616, USA.
Nucleic Acids Res. 2010 Jan;38(3):e13. doi: 10.1093/nar/gkp1012. Epub 2009 Nov 11.
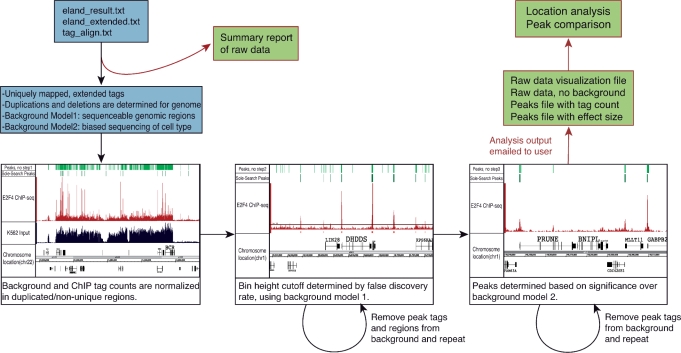
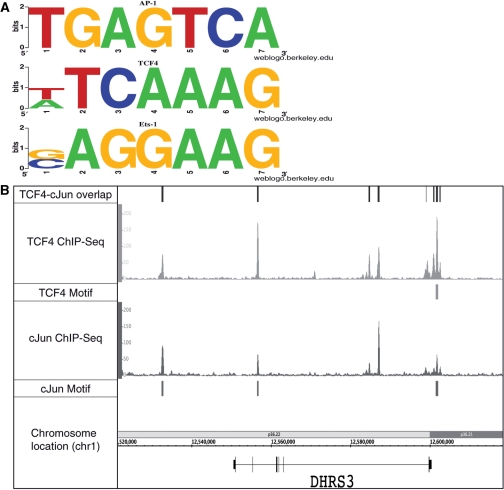
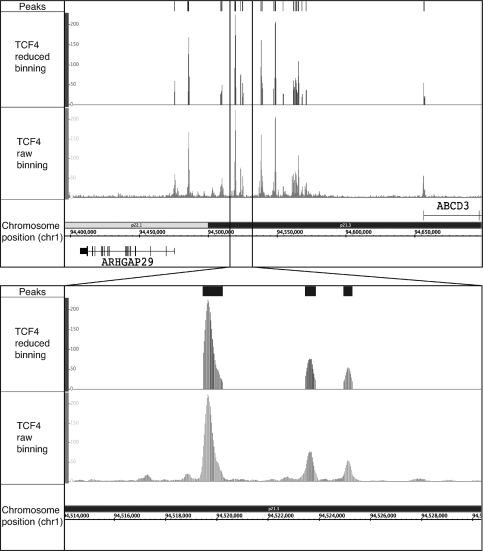
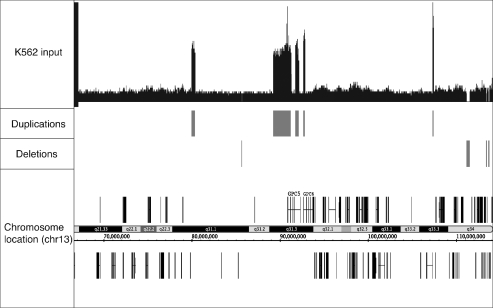
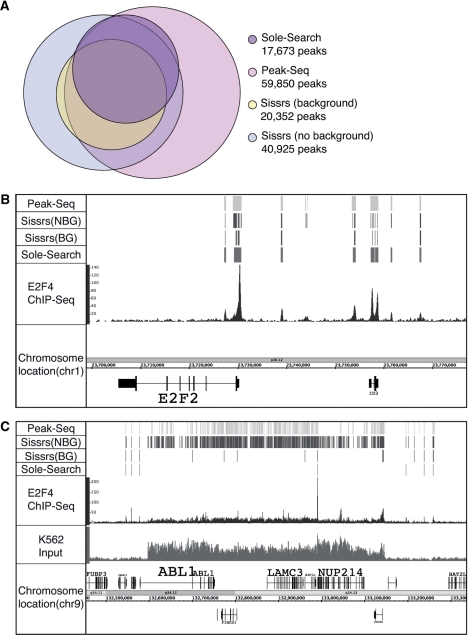
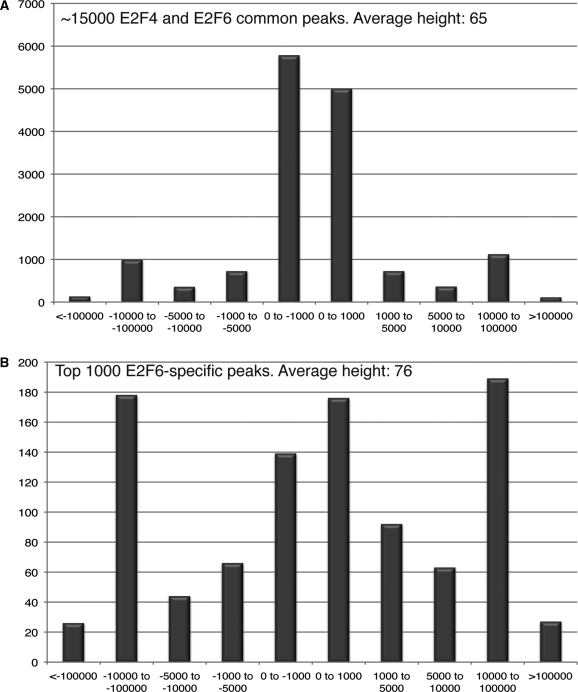
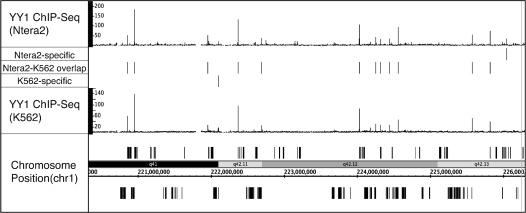
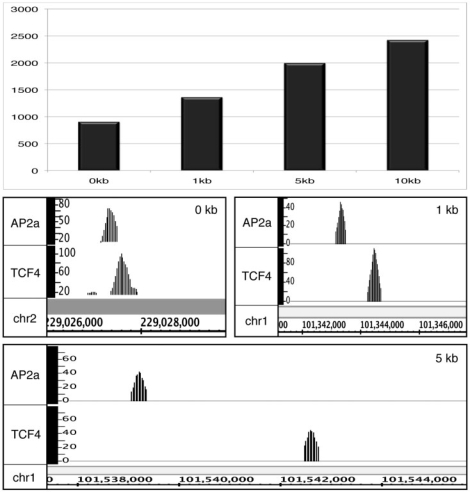
Next-generation sequencing is revolutionizing the identification of transcription factor binding sites throughout the human genome. However, the bioinformatics analysis of large datasets collected using chromatin immunoprecipitation and high-throughput sequencing is often a roadblock that impedes researchers in their attempts to gain biological insights from their experiments. We have developed integrated peak-calling and analysis software (Sole-Search) which is available through a user-friendly interface and (i) converts raw data into a format for visualization on a genome browser, (ii) outputs ranked peak locations using a statistically based method that overcomes the significant problem of false positives, (iii) identifies the gene nearest to each peak, (iv) classifies the location of each peak relative to gene structure, (v) provides information such as the number of binding sites per chromosome and per gene and (vi) allows the user to determine overlap between two different experiments. In addition, the program performs an analysis of amplified and deleted regions of the input genome. This software is web-based and automated, allowing easy and immediate access to all investigators. We demonstrate the utility of our software by collecting, analyzing and comparing ChIP-seq data for six different human transcription factors/cell line combinations.
下一代测序技术正在彻底改变人类基因组中转录因子结合位点的鉴定。然而,使用染色质免疫沉淀和高通量测序收集的大型数据集的生物信息学分析通常是一个障碍,阻碍研究人员从实验中获得生物学见解。我们开发了集成的峰调用和分析软件(Sole-Search),该软件通过用户友好的界面提供,(i)将原始数据转换为可在基因组浏览器上可视化的格式,(ii)使用基于统计学的方法输出排名靠前的峰位置,该方法克服了假阳性的重大问题,(iii)识别每个峰附近的基因,(iv)相对于基因结构对每个峰的位置进行分类,(v)提供有关每个染色体和基因的结合位点数量等信息,以及(vi)允许用户确定两个不同实验之间的重叠。此外,该程序对输入基因组的扩增和缺失区域进行分析。该软件基于网络且自动化,允许所有研究人员轻松、即时地访问。我们通过收集、分析和比较六个不同的人类转录因子/细胞系组合的 ChIP-seq 数据来证明我们软件的实用性。