Department of Computer Science, University of Auckland, New Zealand.
Mol Biol Evol. 2010 Mar;27(3):570-80. doi: 10.1093/molbev/msp274. Epub 2009 Nov 11.
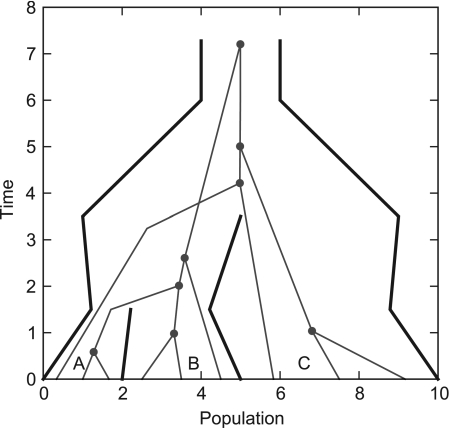
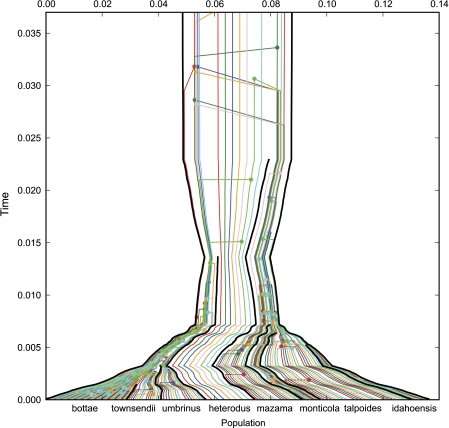
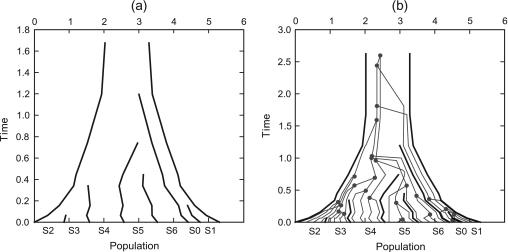
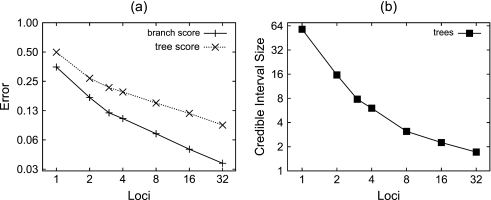
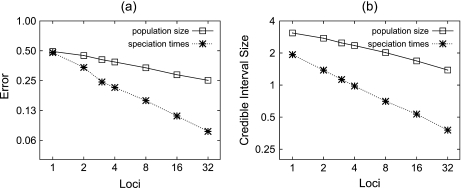
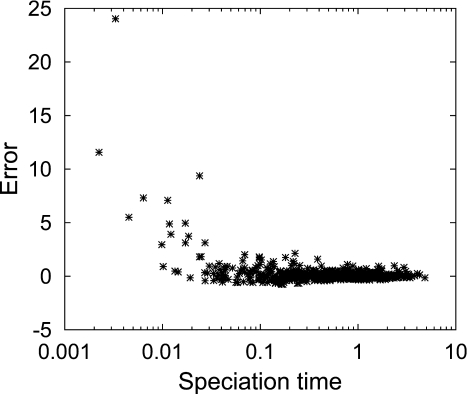
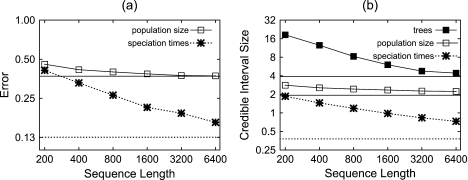
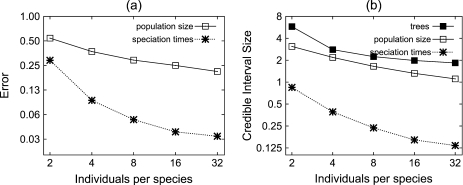
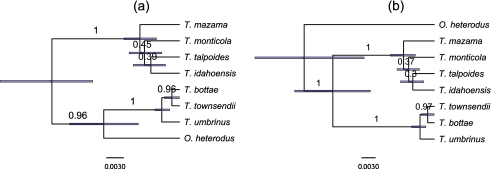
Until recently, it has been common practice for a phylogenetic analysis to use a single gene sequence from a single individual organism as a proxy for an entire species. With technological advances, it is now becoming more common to collect data sets containing multiple gene loci and multiple individuals per species. These data sets often reveal the need to directly model intraspecies polymorphism and incomplete lineage sorting in phylogenetic estimation procedures. For a single species, coalescent theory is widely used in contemporary population genetics to model intraspecific gene trees. Here, we present a Bayesian Markov chain Monte Carlo method for the multispecies coalescent. Our method coestimates multiple gene trees embedded in a shared species tree along with the effective population size of both extant and ancestral species. The inference is made possible by multilocus data from multiple individuals per species. Using a multiindividual data set and a series of simulations of rapid species radiations, we demonstrate the efficacy of our new method. These simulations give some insight into the behavior of the method as a function of sampled individuals, sampled loci, and sequence length. Finally, we compare our new method to both an existing method (BEST 2.2) with similar goals and the supermatrix (concatenation) method. We demonstrate that both BEST and our method have much better estimation accuracy for species tree topology than concatenation, and our method outperforms BEST in divergence time and population size estimation.
直到最近,对系统发育分析来说,使用单个个体的单个基因序列作为整个物种的代表仍然是常见做法。随着技术的进步,现在越来越常见的是收集包含多个基因座和每个物种多个个体的数据组。这些数据集经常需要直接对种内多态性和不完全谱系分选进行建模,以进行系统发育估计程序。对于单一物种,在当代种群遗传学中,合并理论被广泛用于对种内基因树进行建模。在这里,我们提出了一种用于多物种合并的贝叶斯马尔可夫链蒙特卡罗方法。我们的方法同时对嵌入共享物种树中的多个基因树以及现存和祖先物种的有效种群大小进行共估计。这种推断是通过每个物种的多个个体的多基因座数据来实现的。使用多个体数据集和一系列快速物种辐射的模拟,我们证明了我们新方法的有效性。这些模拟深入了解了该方法作为采样个体、采样基因座和序列长度的函数的行为。最后,我们将我们的新方法与具有相似目标的现有方法(BEST 2.2)和超级矩阵(连接)方法进行了比较。我们证明,BEST 和我们的方法在物种树拓扑结构的估计准确性方面都优于连接方法,而我们的方法在分歧时间和种群大小估计方面优于 BEST。