Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN 55905, USA.
BMC Genomics. 2009 Nov 16;10:531. doi: 10.1186/1471-2164-10-531.
Massive parallel sequencing has the potential to replace microarrays as the method for transcriptome profiling. Currently there are two protocols: full-length RNA sequencing (RNA-SEQ) and 3'-tag digital gene expression (DGE). In this preliminary effort, we evaluated the 3' DGE approach using two reference RNA samples from the MicroArray Quality Control Consortium (MAQC).
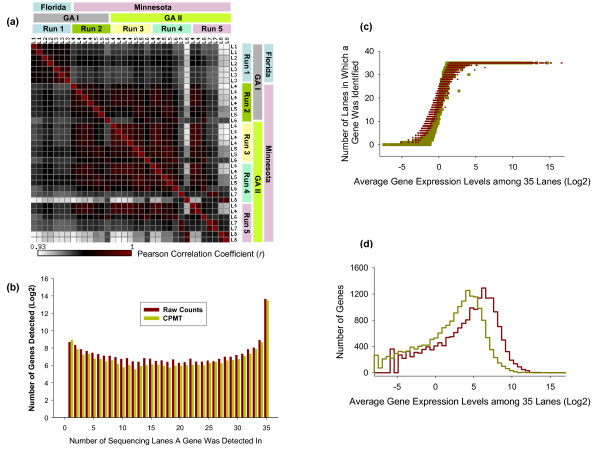
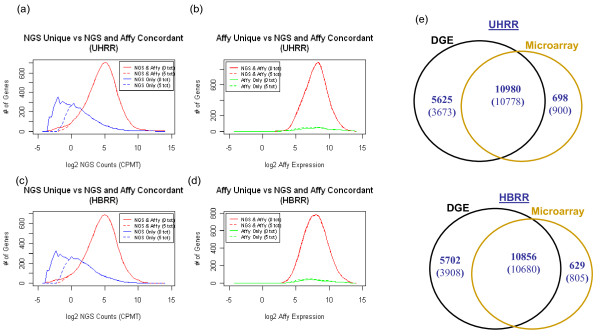
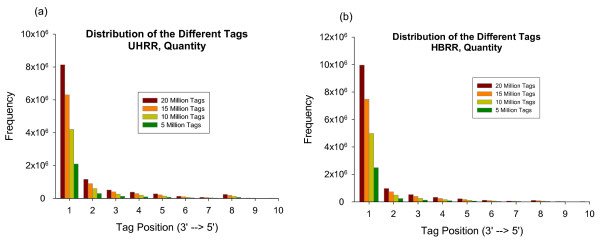
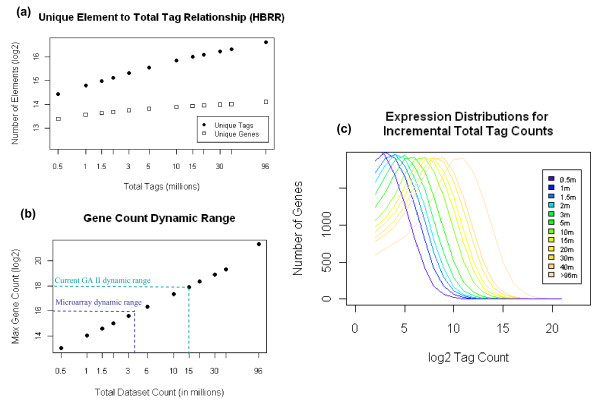
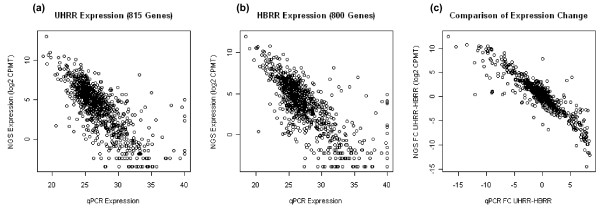
Using Brain RNA sample from multiple runs, we demonstrated that the transcript profiles from 3' DGE were highly reproducible between technical and biological replicates from libraries constructed by the same lab and even by different labs, and between two generations of Illumina's Genome Analyzers. Approximately 65% of all sequence reads mapped to mitochondrial genes, ribosomal RNAs, and canonical transcripts. The expression profiles of brain RNA and universal human reference RNA were compared which demonstrated that DGE was also highly quantitative with excellent correlation of differential expression with quantitative real-time PCR. Furthermore, one lane of 3' DGE sequencing, using the current sequencing chemistry and image processing software, had wider dynamic range for transcriptome profiling and was able to detect lower expressed genes which are normally below the detection threshold of microarrays.
3' tag DGE profiling with massive parallel sequencing achieved high sensitivity and reproducibility for transcriptome profiling. Although it lacks the ability of detecting alternative splicing events compared to RNA-SEQ, it is much more affordable and clearly out-performed microarrays (Affymetrix) in detecting lower abundant transcripts.
大规模平行测序有可能取代微阵列成为转录组分析的方法。目前有两种方案:全长 RNA 测序(RNA-SEQ)和 3'标签数字基因表达(DGE)。在这项初步研究中,我们使用来自微阵列质量控制联盟(MAQC)的两个参考 RNA 样本评估了 3' DGE 方法。
使用来自多个运行的大脑 RNA 样本,我们证明了 3' DGE 的转录谱在由同一实验室甚至不同实验室构建的文库的技术和生物学重复之间以及在两代 Illumina 基因组分析仪之间具有高度可重复性。大约 65%的所有序列读数都映射到线粒体基因、核糖体 RNA 和规范转录本。比较大脑 RNA 和通用人类参考 RNA 的表达谱表明,DGE 也具有高度定量性,差异表达与定量实时 PCR 具有极好的相关性。此外,使用当前测序化学和图像处理软件,3' DGE 测序的一个泳道具有更宽的转录组分析动态范围,并且能够检测到通常低于微阵列检测阈值的低表达基因。
使用大规模平行测序进行 3' 标签 DGE 分析可实现转录组分析的高灵敏度和可重复性。虽然与 RNA-SEQ 相比,它缺乏检测可变剪接事件的能力,但与微阵列(Affymetrix)相比,它的成本要低得多,并且在检测低丰度转录本方面明显优于微阵列。