John Innes Centre, Norwich Research Park, Colney, Norwich, UK.
BMC Genomics. 2009 Nov 18;10:539. doi: 10.1186/1471-2164-10-539.
The Brassica species, related to Arabidopsis thaliana, include an important group of crops and represent an excellent system for studying the evolutionary consequences of polyploidy. Previous studies have led to a proposed structure for an ancestral karyotype and models for the evolution of the B. rapa genome by triplication and segmental rearrangement, but these have not been validated at the sequence level.
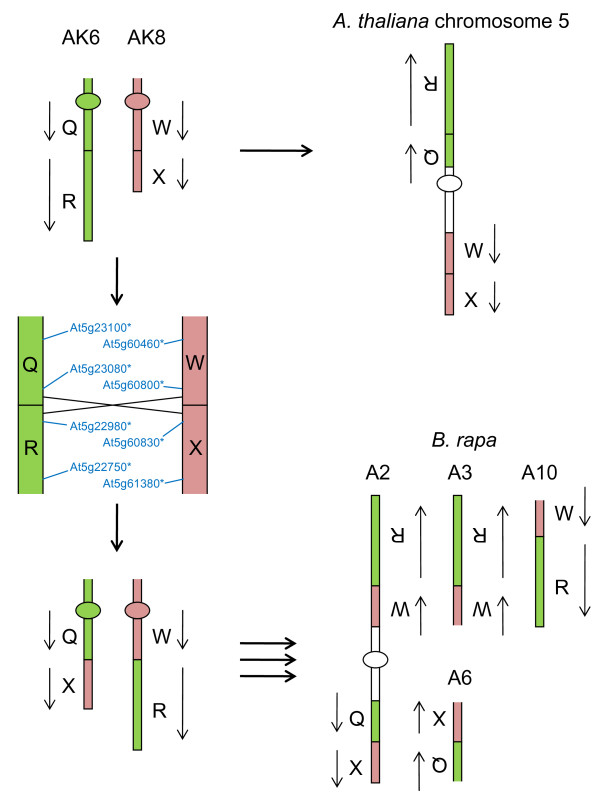
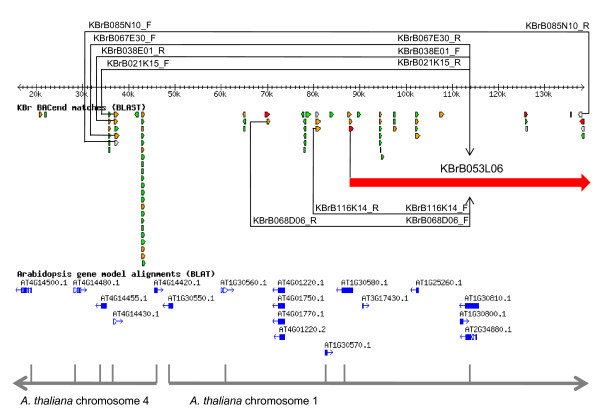
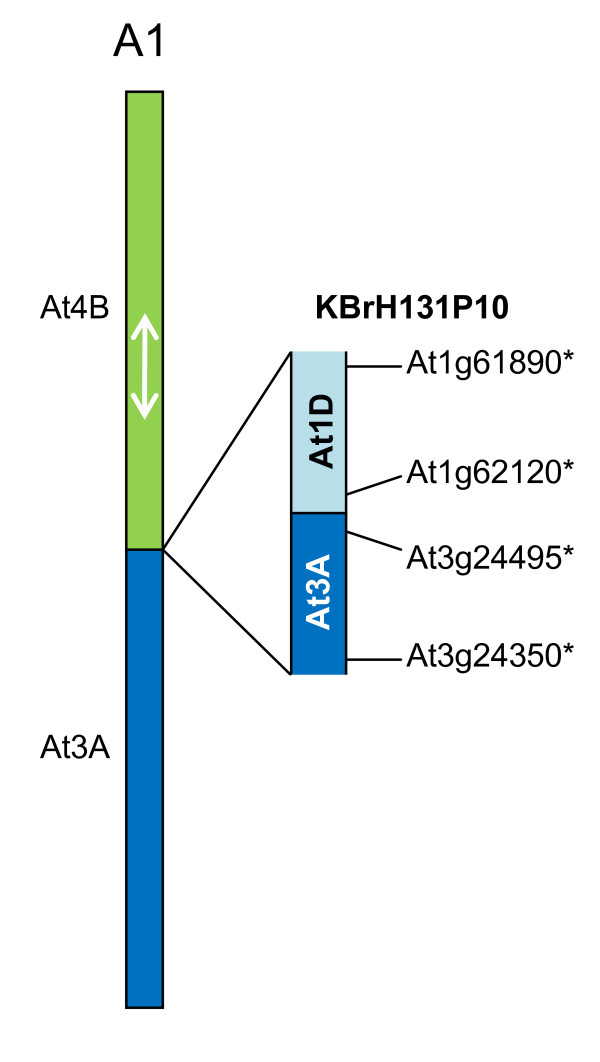
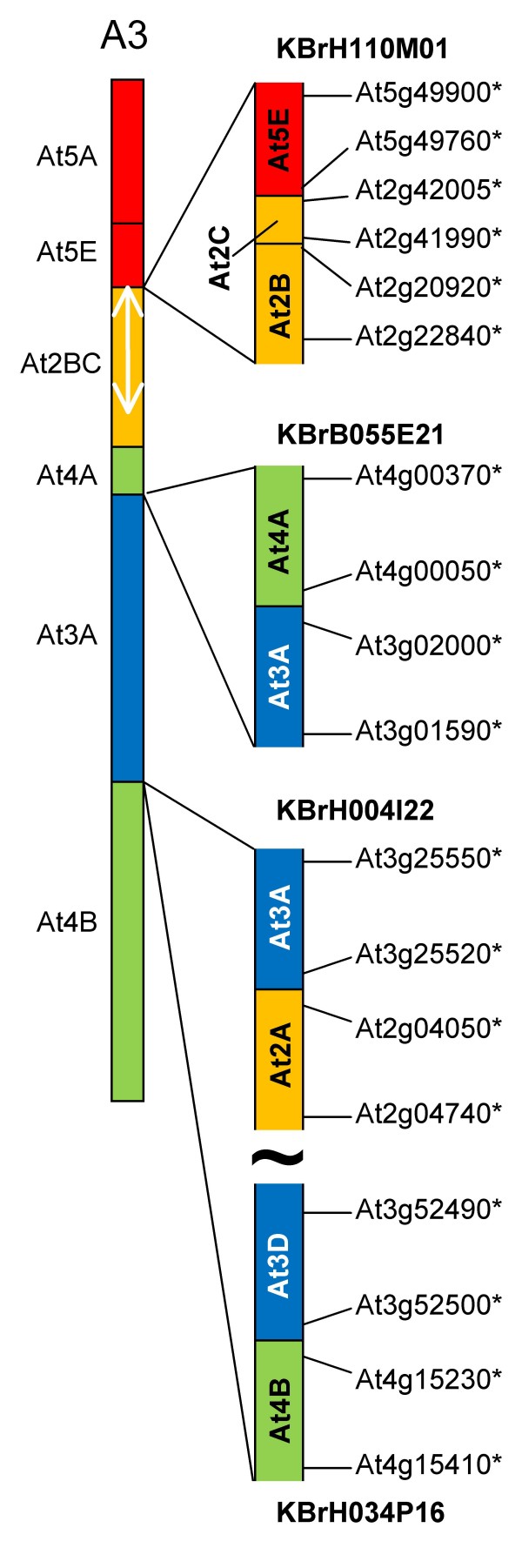
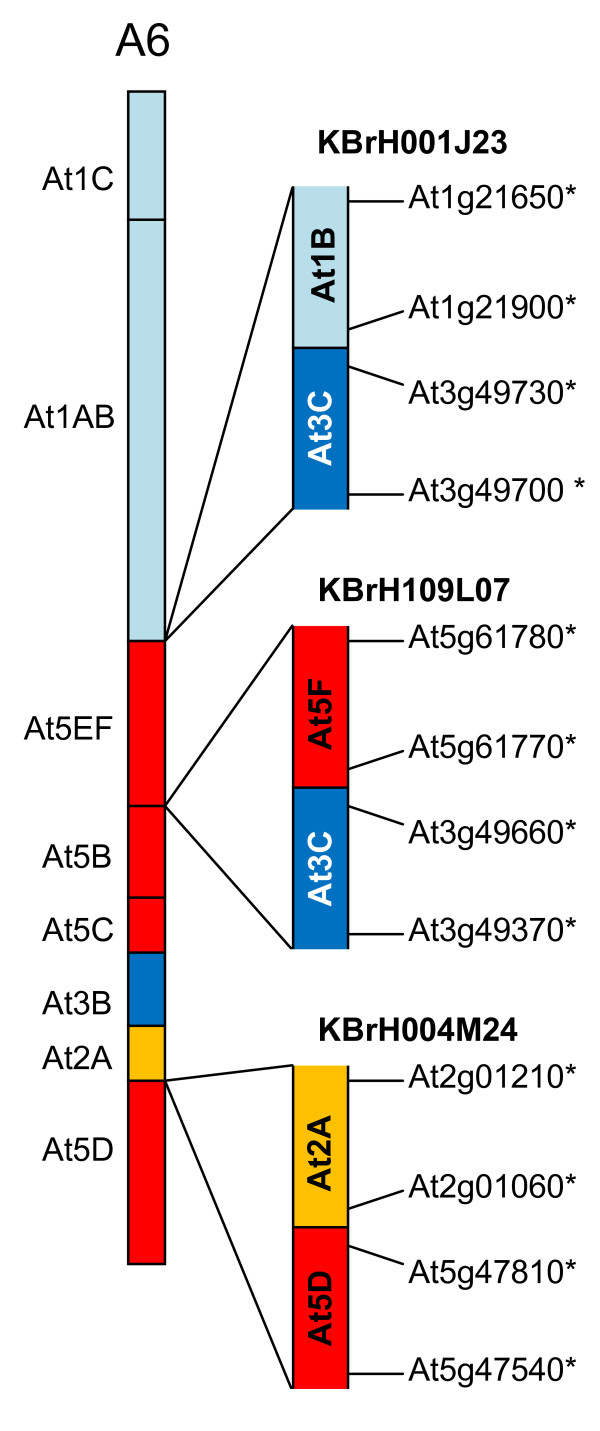
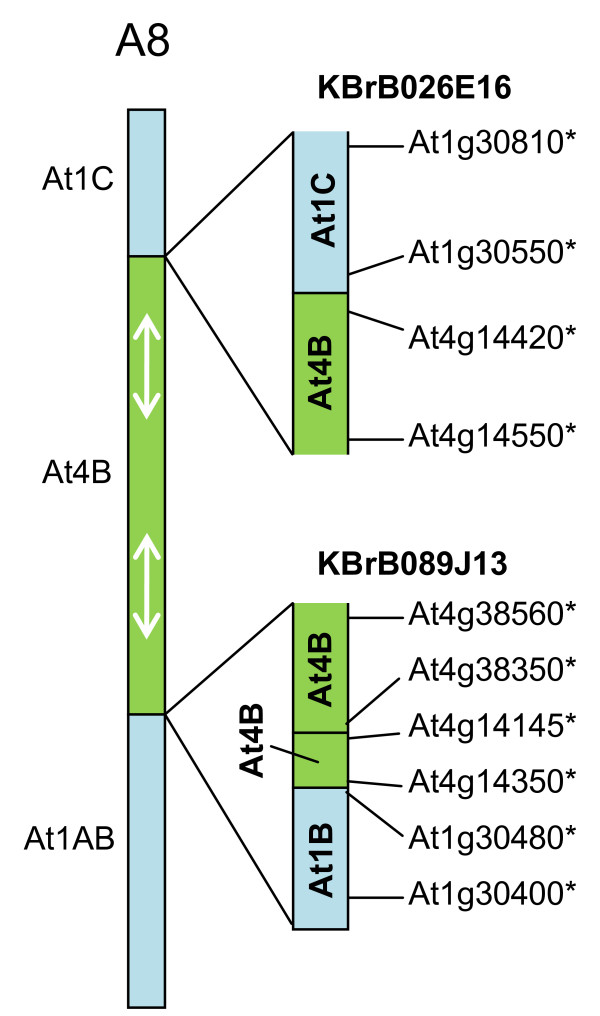
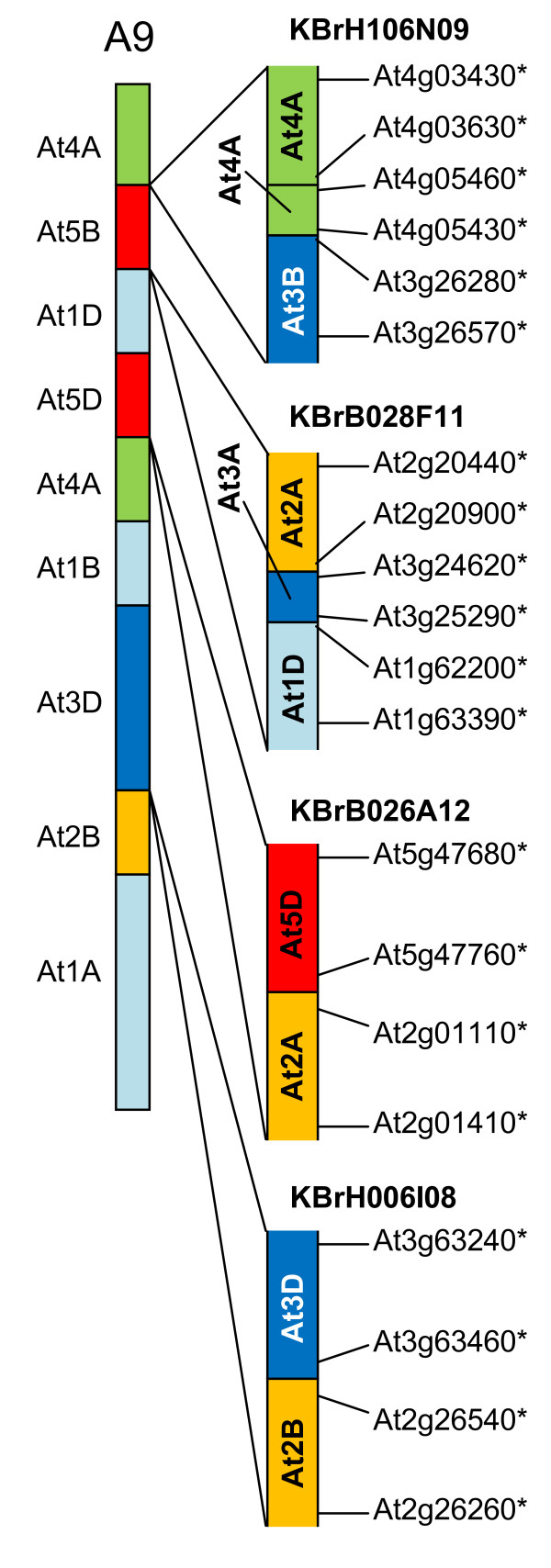
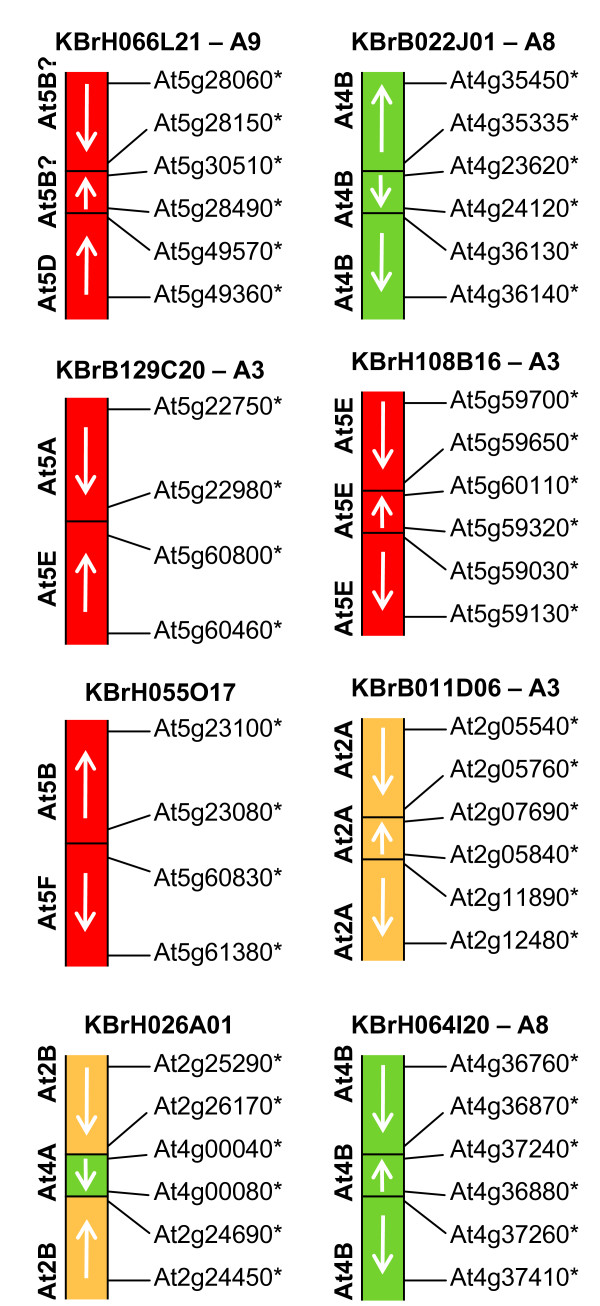
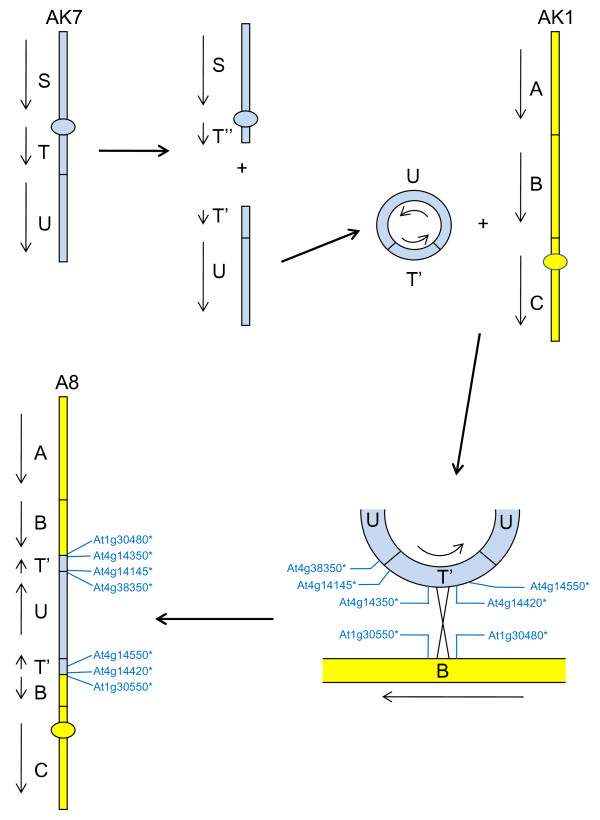
We developed computational tools to analyse the public collection of B. rapa BAC end sequence, in order to identify candidates for representing collinearity discontinuities between the genomes of B. rapa and A. thaliana. For each putative discontinuity, one of the BACs was sequenced and analysed for collinearity with the genome of A. thaliana. Additional BAC clones were identified and sequenced as part of ongoing efforts to sequence four chromosomes of B. rapa. Strikingly few of the 19 inter-chromosomal rearrangements corresponded to the set of collinearity discontinuities anticipated on the basis of previous studies. Our analyses revealed numerous instances of newly detected collinearity blocks. For B. rapa linkage group A8, we were able to develop a model for the derivation of the chromosome from the ancestral karyotype. We were also able to identify a rearrangement event in the ancestor of B. rapa that was not shared with the ancestor of A. thaliana, and is represented in triplicate in the B. rapa genome. In addition to inter-chromosomal rearrangements, we identified and analysed 32 BACs containing the end points of segmental inversion events.
Our results show that previous studies of segmental collinearity between the A. thaliana, Brassica and ancestral karyotype genomes, although very useful, represent over-simplifications of their true relationships. The presence of numerous cryptic collinear genome segments and the frequent occurrence of segmental inversions mean that inference of the positions of genes in B. rapa based on the locations of orthologues in A. thaliana can be misleading. Our results will be of relevance to a wide range of plants that have polyploid genomes, many of which are being considered according to a paradigm of comprising conserved synteny blocks with respect to sequenced, related genomes.
芸薹属植物与拟南芥亲缘关系密切,包括一组重要的作物,是研究多倍体进化后果的理想系统。先前的研究提出了一个祖先核型的结构模型,以及通过三倍体化和片段重排进化的 B. 亚洲甘蓝基因组模型,但这些在序列水平上尚未得到验证。
我们开发了计算工具来分析 B. 亚洲甘蓝 BAC 末端序列的公共集合,以鉴定代表 B. 亚洲甘蓝和 A. 拟南芥基因组之间基因共线性不连续的候选者。对于每个假定的不连续性,我们对其中一个 BAC 进行测序,并分析其与 A. 拟南芥基因组的共线性。作为正在进行的 B. 亚洲甘蓝四条染色体测序工作的一部分,还鉴定和测序了其他 BAC 克隆。令人惊讶的是,19 个染色体间重排中只有少数与先前研究预期的共线性不连续点相对应。我们的分析揭示了许多新检测到的共线性块。对于 B. 亚洲甘蓝连锁群 A8,我们能够从祖先核型推导出该染色体的模型。我们还能够鉴定出 B. 亚洲甘蓝祖先中的一个重排事件,该事件与 A. 拟南芥的祖先不同,并且在 B. 亚洲甘蓝基因组中以三倍体形式存在。除了染色体间重排,我们还鉴定和分析了 32 个包含片段倒位事件终点的 BAC。
我们的结果表明,先前关于 A. 拟南芥、芸薹属和祖先核型基因组之间片段共线性的研究虽然非常有用,但代表了它们真实关系的过度简化。大量隐藏的共线性基因组片段的存在以及片段倒位的频繁发生意味着,根据 A. 拟南芥中同源基因的位置推断 B. 亚洲甘蓝中基因的位置可能会产生误导。我们的研究结果将与许多具有多倍体基因组的植物相关,其中许多植物正在根据与测序相关基因组的保守同线性块范式进行考虑。