Department of Medical Epidemiology, Karolinska Institutet, Stockholm, Sweden.
PLoS One. 2009 Dec 2;4(12):e7969. doi: 10.1371/journal.pone.0007969.
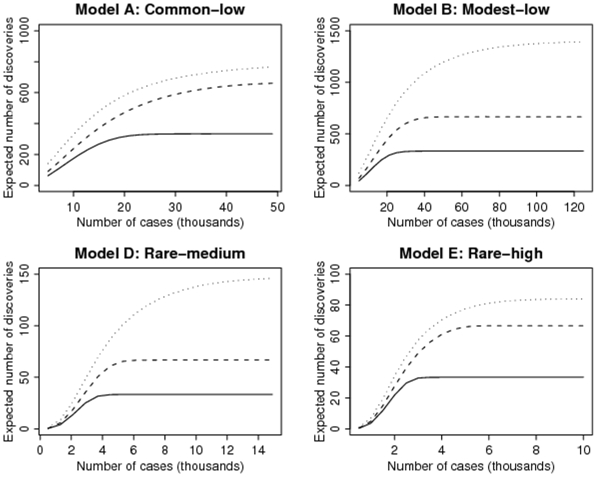
A great majority of genetic markers discovered in recent genome-wide association studies have small effect sizes, and they explain only a small fraction of the genetic contribution to the diseases. How many more variants can we expect to discover and what study sizes are needed? We derive the connection between the cumulative risk of the SNP variants to the latent genetic risk model and heritability of the disease. We determine the sample size required for case-control studies in order to achieve a certain expected number of discoveries in a collection of most significant SNPs. Assuming similar allele frequencies and effect sizes of the currently validated SNPs, complex phenotypes such as type-2 diabetes would need approximately 800 variants to explain its 40% heritability. Much smaller numbers of variants are needed if we assume rare-variants but higher penetrance models. We estimate that up to 50,000 cases and an equal number of controls are needed to discover 800 common low-penetrant variants among the top 5000 SNPs. Under common and rare low-penetrance models, the very large studies required to discover the numerous variants are probably at the limit of practical feasibility. Under rare-variant with medium- to high-penetrance models (odds-ratios between 1.6 and 4.0), studies comparable in size to many existing studies are adequate provided the genotyping technology can interrogate more and rarer variants.
绝大多数在最近的全基因组关联研究中发现的遗传标记具有较小的效应大小,并且仅解释了疾病遗传贡献的一小部分。我们可以期望发现多少更多的变体,以及需要多大的研究规模?我们推导出 SNP 变体的累积风险与潜在遗传风险模型和疾病遗传性之间的联系。我们确定了病例对照研究所需的样本量,以便在一组最重要的 SNP 中达到一定数量的预期发现。假设目前已验证的 SNP 的等位基因频率和效应大小相似,那么像 2 型糖尿病这样的复杂表型需要大约 800 个变体才能解释其 40%的遗传性。如果我们假设稀有变体但具有更高外显率的模型,则需要的变体数量要少得多。我们估计,需要多达 50,000 例病例和相同数量的对照,才能在排名前 5,000 的 SNP 中发现 800 个常见的低外显率变体。在常见和稀有低外显率模型下,发现大量常见低外显率变体所需的非常大规模的研究可能已经达到了实际可行性的极限。在罕见变体且具有中到高外显率的模型(比值比在 1.6 到 4.0 之间)下,如果基因分型技术能够检测到更多和更罕见的变体,则与许多现有研究规模相当的研究就足够了。