Department of Food Safety, Norwegian School of Veterinary Science, Oslo, Norway.
PLoS One. 2009 Dec 2;4(12):e8113. doi: 10.1371/journal.pone.0008113.
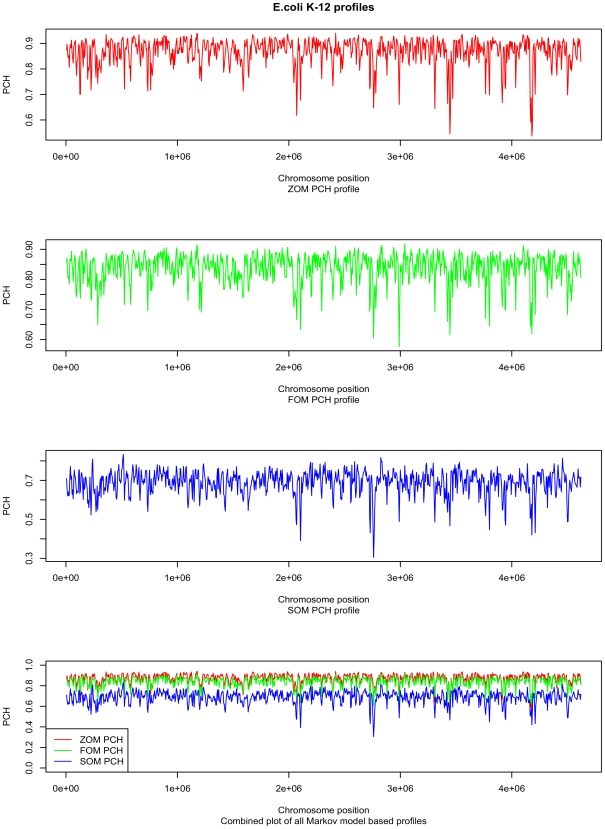
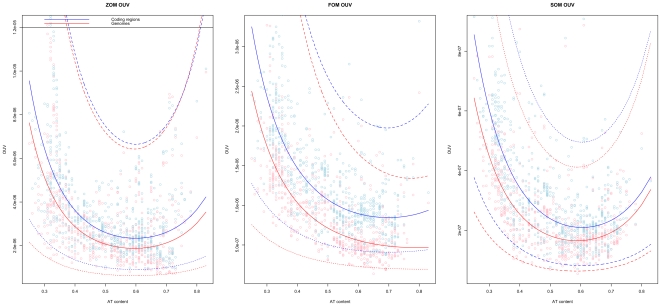
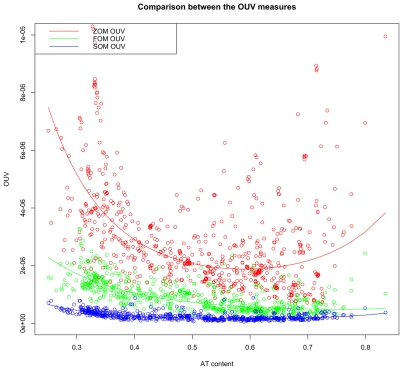
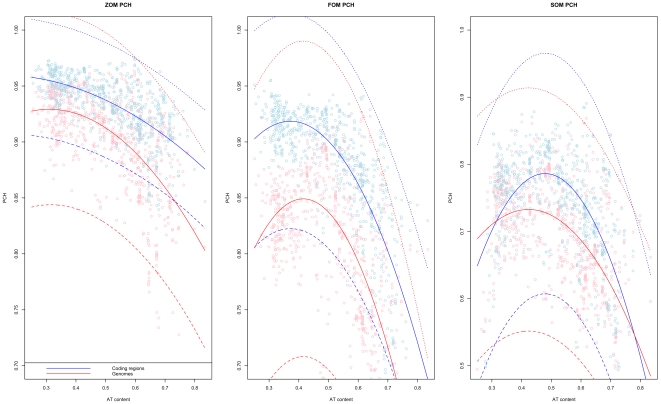
DNA word frequencies, normalized for genomic AT content, are remarkably stable within prokaryotic genomes and are therefore said to reflect a "genomic signature." The genomic signatures can be used to phylogenetically classify organisms from arbitrary sampled DNA. Genomic signatures can also be used to search for horizontally transferred DNA or DNA regions subjected to special selection forces. Thus, the stability of the genomic signature can be used as a measure of genomic homogeneity. The factors associated with the stability of the genomic signatures are not known, and this motivated us to investigate further. We analyzed the intra-genomic variance of genomic signatures based on AT content normalization (0(th) order Markov model) as well as genomic signatures normalized by smaller DNA words (1(st) and 2(nd) order Markov models) for 636 sequenced prokaryotic genomes. Regression models were fitted, with intra-genomic signature variance as the response variable, to a set of factors representing genomic properties such as genomic AT content, genome size, habitat, phylum, oxygen requirement, optimal growth temperature and oligonucleotide usage variance (OUV, a measure of oligonucleotide usage bias), measured as the variance between genomic tetranucleotide frequencies and Markov chain approximated tetranucleotide frequencies, as predictors.
Regression analysis revealed that OUV was the most important factor (p<0.001) determining intra-genomic homogeneity as measured using genomic signatures. This means that the less random the oligonucleotide usage is in the sense of higher OUV, the more homogeneous the genome is in terms of the genomic signature. The other factors influencing variance in the genomic signature (p<0.001) were genomic AT content, phylum and oxygen requirement.
Genomic homogeneity in prokaryotes is intimately linked to genomic GC content, oligonucleotide usage bias (OUV) and aerobiosis, while oligonucleotide usage bias (OUV) is associated with genomic GC content, aerobiosis and habitat.
经基因组 AT 含量归一化后的 DNA 单词频率在原核基因组中非常稳定,因此被认为反映了一种“基因组特征”。基因组特征可用于对任意采样 DNA 进行系统发育分类的生物体。基因组特征也可用于搜索水平转移的 DNA 或受特殊选择压力影响的 DNA 区域。因此,基因组特征的稳定性可用作基因组同质性的衡量标准。与基因组特征稳定性相关的因素尚不清楚,这促使我们进行了进一步的研究。我们基于 AT 含量归一化(0 阶马尔可夫模型)以及通过较小的 DNA 单词归一化的基因组特征(1 阶和 2 阶马尔可夫模型),分析了 636 个已测序原核基因组的基因组特征内的方差。以基因组特征内方差作为响应变量,针对一系列代表基因组特性的因素(包括基因组 AT 含量、基因组大小、栖息地、门、氧气需求、最佳生长温度和寡核苷酸使用方差[OUV,衡量寡核苷酸使用偏向的指标,作为基因组四核苷酸频率和马尔可夫链逼近四核苷酸频率之间的方差])拟合回归模型。
回归分析表明,寡核苷酸使用方差(OUV)是决定使用基因组特征衡量的基因组内同质性的最重要因素(p<0.001)。这意味着,寡核苷酸使用的随机性越低,即 OUV 越高,基因组在基因组特征方面的同质性就越高。影响基因组特征方差的其他因素(p<0.001)包括基因组 AT 含量、门和氧气需求。
原核生物的基因组同质性与基因组 GC 含量、寡核苷酸使用偏向(OUV)和需氧性密切相关,而寡核苷酸使用偏向(OUV)与基因组 GC 含量、需氧性和栖息地相关。