State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing 100193, China.
BMC Bioinformatics. 2009 Dec 14;10:416. doi: 10.1186/1471-2105-10-416.
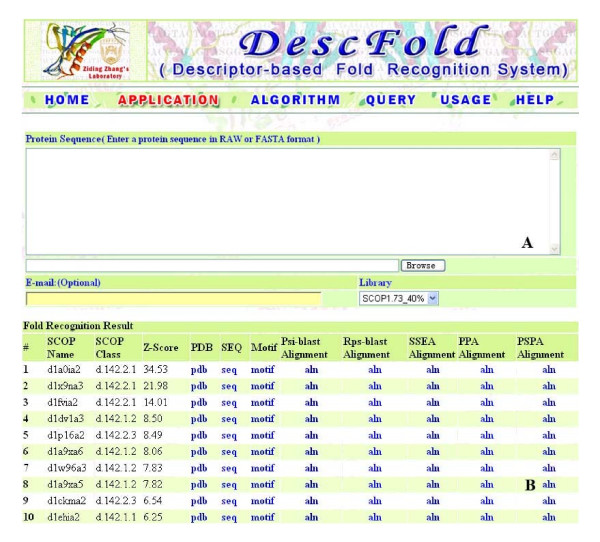
Machine learning-based methods have been proven to be powerful in developing new fold recognition tools. In our previous work [Zhang, Kochhar and Grigorov (2005) Protein Science, 14: 431-444], a machine learning-based method called DescFold was established by using Support Vector Machines (SVMs) to combine the following four descriptors: a profile-sequence-alignment-based descriptor using Psi-blast e-values and bit scores, a sequence-profile-alignment-based descriptor using Rps-blast e-values and bit scores, a descriptor based on secondary structure element alignment (SSEA), and a descriptor based on the occurrence of PROSITE functional motifs. In this work, we focus on the improvement of DescFold by incorporating more powerful descriptors and setting up a user-friendly web server.
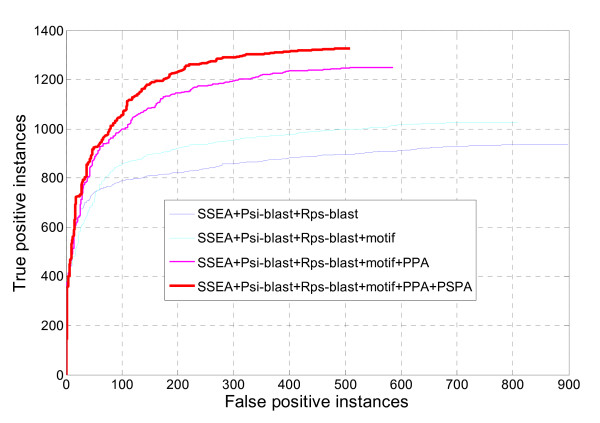
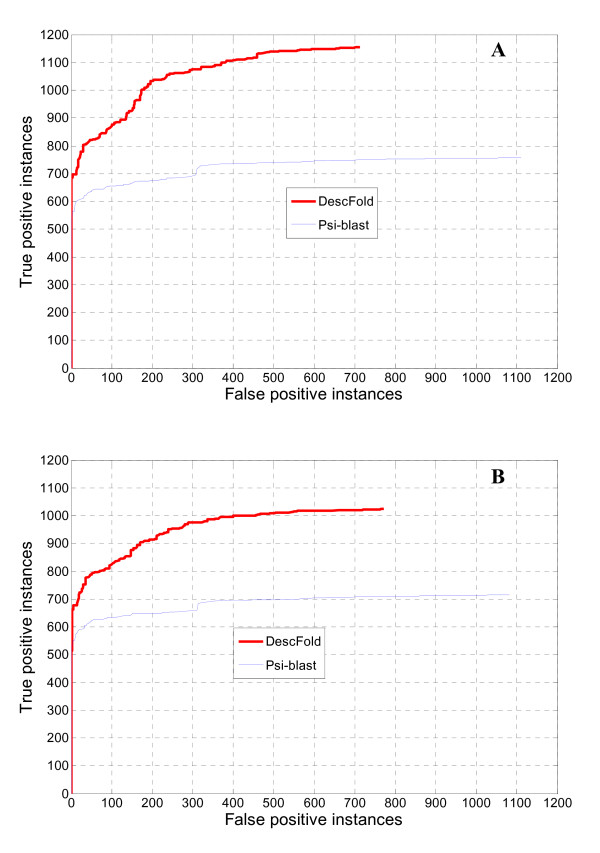
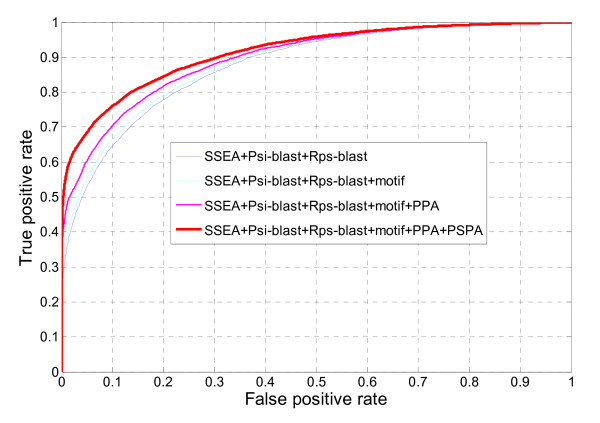
In seeking more powerful descriptors, the profile-profile alignment score generated from the COMPASS algorithm was first considered as a new descriptor (i.e., PPA). When considering a profile-profile alignment between two proteins in the context of fold recognition, one protein is regarded as a template (i.e., its 3D structure is known). Instead of a sequence profile derived from a Psi-blast search, a structure-seeded profile for the template protein was generated by searching its structural neighbors with the assistance of the TM-align structural alignment algorithm. Moreover, the COMPASS algorithm was used again to derive a profile-structural-profile-alignment-based descriptor (i.e., PSPA). We trained and tested the new DescFold in a total of 1,835 highly diverse proteins extracted from the SCOP 1.73 version. When the PPA and PSPA descriptors were introduced, the new DescFold boosts the performance of fold recognition substantially. Using the SCOP_1.73_40% dataset as the fold library, the DescFold web server based on the trained SVM models was further constructed. To provide a large-scale test for the new DescFold, a stringent test set of 1,866 proteins were selected from the SCOP 1.75 version. At a less than 5% false positive rate control, the new DescFold is able to correctly recognize structural homologs at the fold level for nearly 46% test proteins. Additionally, we also benchmarked the DescFold method against several well-established fold recognition algorithms through the LiveBench targets and Lindahl dataset.
The new DescFold method was intensively benchmarked to have very competitive performance compared with some well-established fold recognition methods, suggesting that it can serve as a useful tool to assist in template-based protein structure prediction. The DescFold server is freely accessible at http://202.112.170.199/DescFold/index.html.
基于机器学习的方法已被证明在开发新的折叠识别工具方面非常有效。在我们之前的工作中[Zhang、Kochhar 和 Grigorov(2005)《蛋白质科学》,14:431-444],我们使用支持向量机(SVM)建立了一种基于机器学习的方法 DescFold,该方法结合了以下四个描述符:基于 Psi-blast e 值和位得分的序列-结构比对描述符、基于 Rps-blast e 值和位得分的结构-序列比对描述符、基于二级结构元素比对(SSEA)的描述符以及基于 PROSITE 功能基序的描述符。在这项工作中,我们专注于通过结合更强大的描述符并建立一个用户友好的网络服务器来改进 DescFold。
为了寻求更强大的描述符,我们首先考虑了 COMPASS 算法生成的结构-结构比对得分作为新的描述符(即 PPA)。在折叠识别的背景下考虑两个蛋白质之间的结构-结构比对时,一个蛋白质被视为模板(即其 3D 结构是已知的)。而不是从 Psi-blast 搜索中获得序列结构,而是通过 TM-align 结构比对算法搜索其结构邻居,为模板蛋白生成基于结构的结构谱。此外,我们再次使用 COMPASS 算法来获得基于结构-结构-结构比对的描述符(即 PSPA)。我们在总共 1835 种高度多样化的蛋白质中对新的 DescFold 进行了训练和测试,这些蛋白质是从 SCOP 1.73 版本中提取的。当引入 PPA 和 PSPA 描述符时,新的 DescFold 大大提高了折叠识别的性能。使用 SCOP_1.73_40%数据集作为折叠库,我们进一步构建了基于训练的 SVM 模型的 DescFold 网络服务器。为了对新的 DescFold 进行大规模测试,我们从 SCOP 1.75 版本中选择了 1866 种蛋白质作为严格测试集。在控制假阳性率低于 5%的情况下,新的 DescFold 能够正确识别结构同源物,约有近 46%的测试蛋白质属于折叠水平。此外,我们还通过 LiveBench 目标和 Lindahl 数据集将 DescFold 方法与几个成熟的折叠识别算法进行了基准测试。
新的 DescFold 方法经过了密集的基准测试,与一些成熟的折叠识别方法相比具有非常有竞争力的性能,这表明它可以作为一种有用的工具,辅助基于模板的蛋白质结构预测。DescFold 服务器可在 http://202.112.170.199/DescFold/index.html 免费访问。