Institute of Cellular and Molecular Biology (MBB 3 210B), Center for Systems and Synthetic Biology, University of Texas at Austin, 2500 Speedway, Austin, Texas, USA.
BMC Bioinformatics. 2009 Dec 14;10:419. doi: 10.1186/1471-2105-10-419.
Protein-protein interactions underlie many important biological processes. Computational prediction methods can nicely complement experimental approaches for identifying protein-protein interactions. Recently, a unique category of sequence-based prediction methods has been put forward--unique in the sense that it does not require homologous protein sequences. This enables it to be universally applicable to all protein sequences unlike many of previous sequence-based prediction methods. If effective as claimed, these new sequence-based, universally applicable prediction methods would have far-reaching utilities in many areas of biology research.
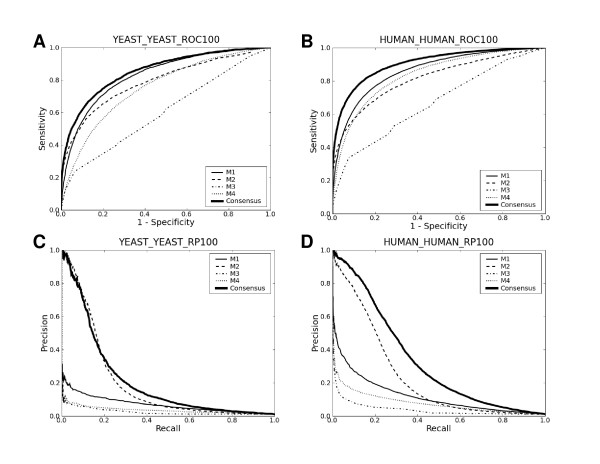
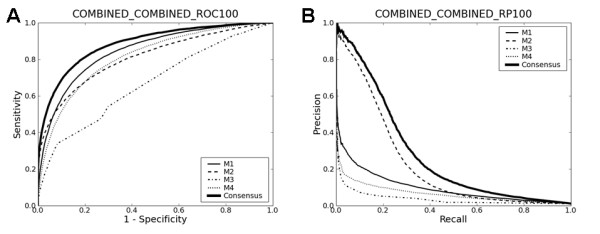
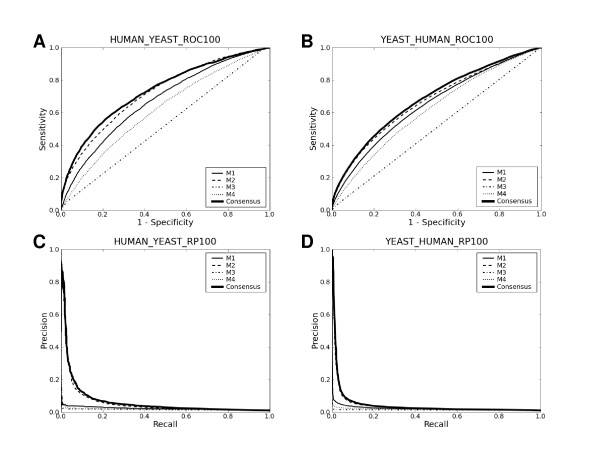
Upon close survey, I realized that many of these new methods were ill-tested. In addition, newer methods were often published without performance comparison with previous ones. Thus, it is not clear how good they are and whether there are significant performance differences among them. In this study, I have implemented and thoroughly tested 4 different methods on large-scale, non-redundant data sets. It reveals several important points. First, significant performance differences are noted among different methods. Second, data sets typically used for training prediction methods appear significantly biased, limiting the general applicability of prediction methods trained with them. Third, there is still ample room for further developments. In addition, my analysis illustrates the importance of complementary performance measures coupled with right-sized data sets for meaningful benchmark tests.
The current study reveals the potentials and limits of the new category of sequence-based protein-protein interaction prediction methods, which in turn provides a firm ground for future endeavours in this important area of contemporary bioinformatics.
蛋白质-蛋白质相互作用是许多重要生物过程的基础。计算预测方法可以很好地补充识别蛋白质-蛋白质相互作用的实验方法。最近,提出了一种独特的基于序列的预测方法类别——独特之处在于它不需要同源蛋白质序列。这使得它能够普遍适用于所有蛋白质序列,不像许多以前的基于序列的预测方法。如果如声称的那样有效,这些新的基于序列的、普遍适用的预测方法将在生物学研究的许多领域具有深远的应用价值。
经过仔细调查,我意识到这些新方法中有许多未经充分测试。此外,新方法通常在没有与以前的方法进行性能比较的情况下发布。因此,不清楚它们的效果如何,以及它们之间是否存在显著的性能差异。在这项研究中,我在大规模、非冗余数据集上实现并彻底测试了 4 种不同的方法。它揭示了几个重要的观点。首先,不同方法之间存在显著的性能差异。其次,用于训练预测方法的数据集通常存在明显的偏差,限制了使用它们训练的预测方法的普遍适用性。第三,还有很大的发展空间。此外,我的分析说明了互补性能指标与合适大小的数据集相结合对于有意义的基准测试的重要性。
本研究揭示了新型基于序列的蛋白质-蛋白质相互作用预测方法的潜力和局限性,这反过来为当代生物信息学这一重要领域的未来努力提供了坚实的基础。