Department of Medical Informatics and Biomathematics, University of Münster, Domagkstrasse 9, 48149 Münster, Germany.
BMC Bioinformatics. 2009 Dec 15;10:422. doi: 10.1186/1471-2105-10-422.
Multiple gene expression signatures derived from microarray experiments have been published in the field of leukemia research. A comparison of these signatures with results from new experiments is useful for verification as well as for interpretation of the results obtained. Currently, the percentage of overlapping genes is frequently used to compare published gene signatures against a signature derived from a new experiment. However, it has been shown that the percentage of overlapping genes is of limited use for comparing two experiments due to the variability of gene signatures caused by different array platforms or assay-specific influencing parameters. Here, we present a robust approach for a systematic and quantitative comparison of published gene expression signatures with an exemplary query dataset.
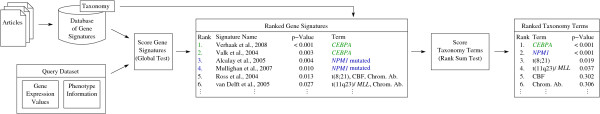
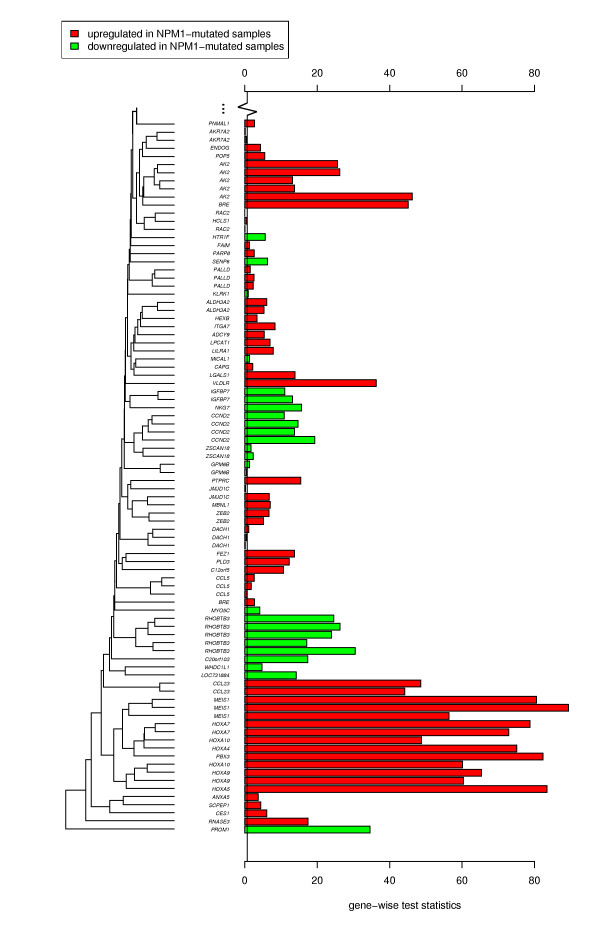
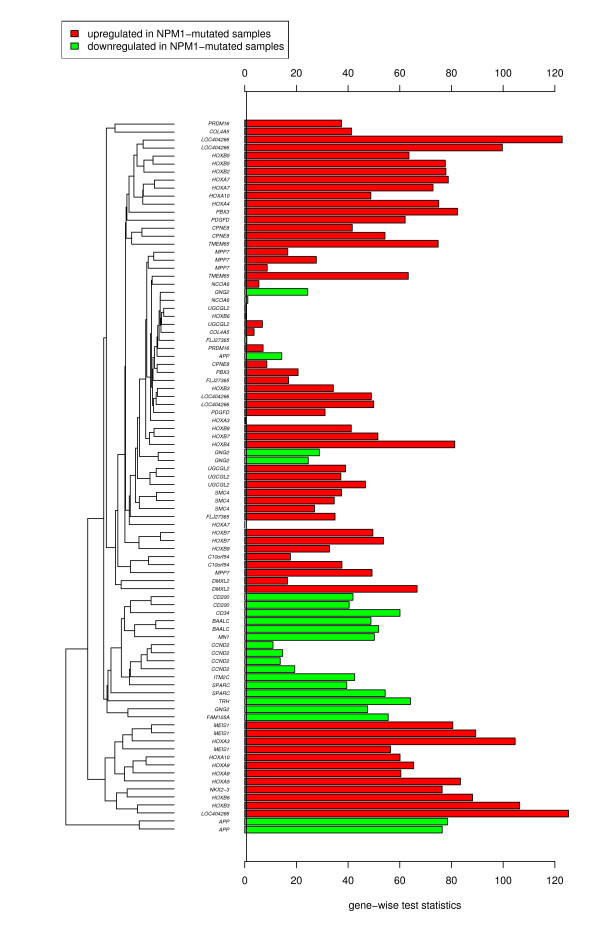
A database storing 138 leukemia-related published gene signatures was designed. Each gene signature was manually annotated with terms according to a leukemia-specific taxonomy. Two analysis steps are implemented to compare a new microarray dataset with the results from previous experiments stored and curated in the database. First, the global test method is applied to assess gene signatures and to constitute a ranking among them. In a subsequent analysis step, the focus is shifted from single gene signatures to chromosomal aberrations or molecular mutations as modeled in the taxonomy. Potentially interesting disease characteristics are detected based on the ranking of gene signatures associated with these aberrations stored in the database. Two example analyses are presented. An implementation of the approach is freely available as web-based application.
The presented approach helps researchers to systematically integrate the knowledge derived from numerous microarray experiments into the analysis of a new dataset. By means of example leukemia datasets we demonstrate that this approach detects related experiments as well as related molecular mutations and may help to interpret new microarray data.
来自微阵列实验的多个基因表达特征已在白血病研究领域中发表。将这些特征与新实验的结果进行比较,对于验证和解释获得的结果都是有用的。目前,经常使用重叠基因的百分比来比较来自新实验的特征与已发表的基因特征。然而,由于不同的阵列平台或特定于检测的影响参数引起的基因特征的可变性,已经表明重叠基因的百分比对于比较两个实验的用途有限。在这里,我们提出了一种稳健的方法,用于系统地和定量地将已发表的基因表达特征与示例查询数据集进行比较。
设计了一个存储 138 个与白血病相关的已发表基因特征的数据库。根据白血病特定的分类法,每个基因特征都用术语进行手动注释。实现了两个分析步骤来比较新的微阵列数据集与存储在数据库中的以前的实验结果。首先,应用全局测试方法来评估基因特征并在它们之间构成排名。在随后的分析步骤中,重点从单个基因特征转移到分类法中建模的染色体异常或分子突变。基于与这些异常相关的存储在数据库中的基因特征的排名,检测潜在的有趣疾病特征。呈现了两个示例分析。该方法的实现可作为基于网络的应用程序免费获得。
所提出的方法有助于研究人员将从众多微阵列实验中获得的知识系统地整合到新数据集的分析中。通过示例白血病数据集,我们证明了该方法可以检测到相关的实验以及相关的分子突变,并有助于解释新的微阵列数据。