Department of Immunology and Molecular Pathology, Royal Free Hospital and University College London, London, UK.
J Allergy Clin Immunol. 2009 Dec;124(6):1289-302.e4. doi: 10.1016/j.jaci.2009.10.038.
The genetic etiologies of the hyper-IgE syndromes are diverse. Approximately 60% to 70% of patients with hyper-IgE syndrome have dominant mutations in STAT3, and a single patient was reported to have a homozygous TYK2 mutation. In the remaining patients with hyper-IgE syndrome, the genetic etiology has not yet been identified.
We aimed to identify a gene that is mutated or deleted in autosomal recessive hyper-IgE syndrome.
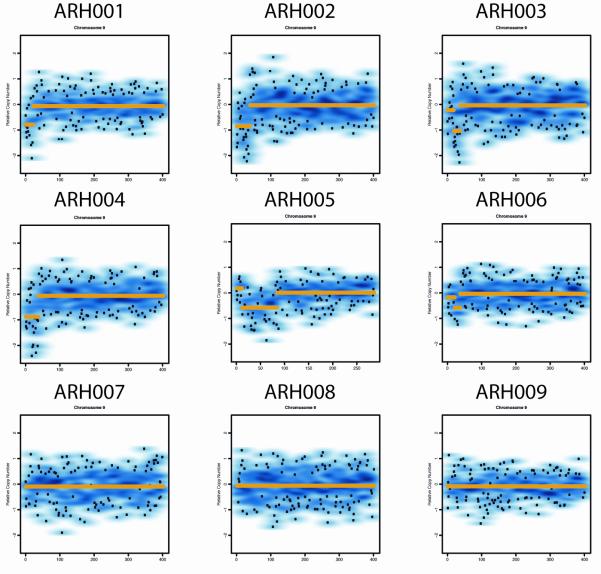
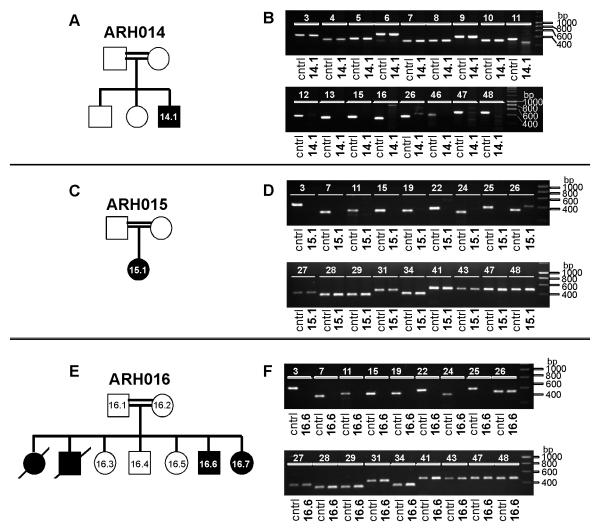
We performed genome-wide single nucleotide polymorphism analysis for 9 patients with autosomal-recessive hyper-IgE syndrome to locate copy number variations and homozygous haplotypes. Homozygosity mapping was performed with 12 patients from 7 additional families. The candidate gene was analyzed by genomic and cDNA sequencing to identify causative alleles in a total of 27 patients with autosomal-recessive hyper-IgE syndrome.
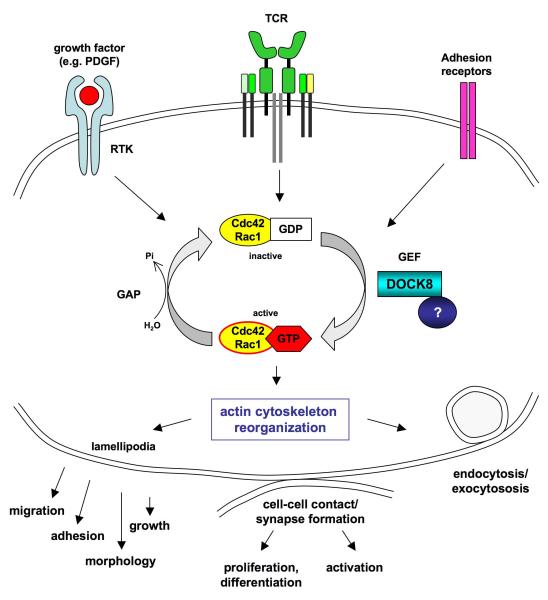
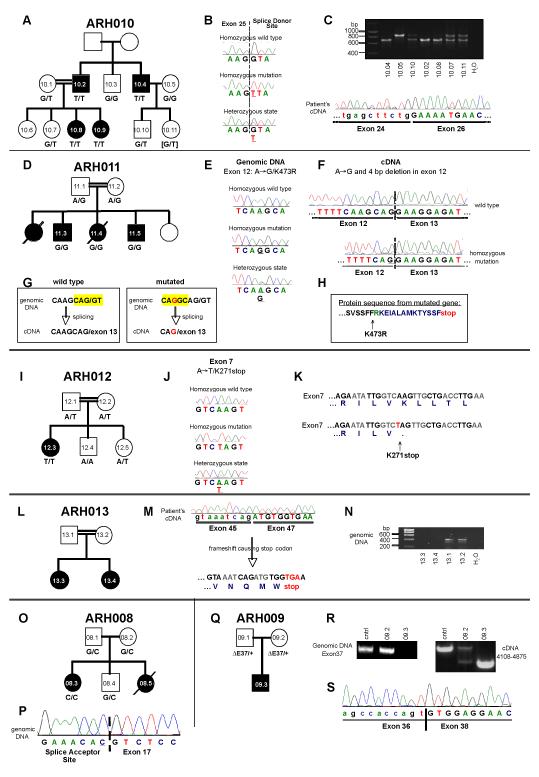
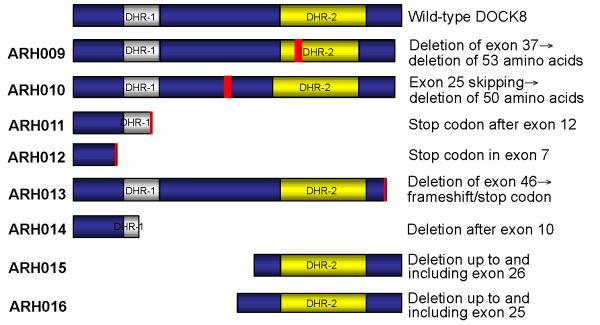
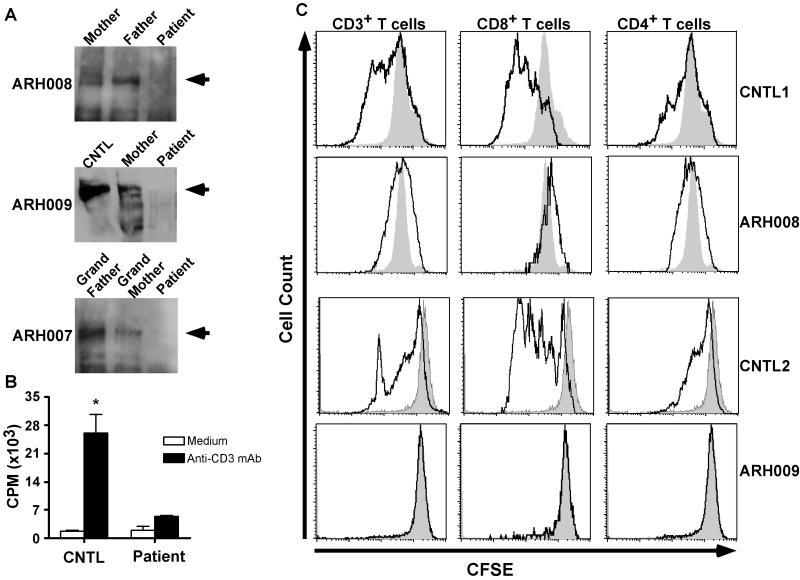
Subtelomeric biallelic microdeletions were identified in 5 patients at the terminus of chromosome 9p. In all 5 patients, the deleted interval involved dedicator of cytokinesis 8 (DOCK8), encoding a protein implicated in the regulation of the actin cytoskeleton. Sequencing of patients without large deletions revealed 16 patients from 9 unrelated families with distinct homozygous mutations in DOCK8 causing premature termination, frameshift, splice site disruption, and single exon deletions and microdeletions. DOCK8 deficiency was associated with impaired activation of CD4+ and CD8+T cells.
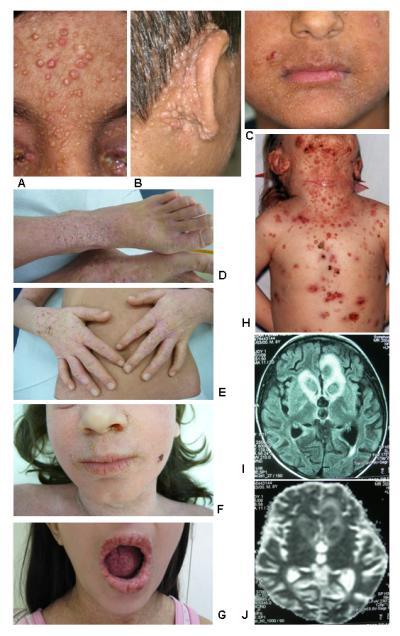
Autosomal-recessive mutations in DOCK8 are responsible for many, although not all, cases of autosomal-recessive hyper-IgE syndrome. DOCK8 disruption is associated with a phenotype of severe cellular immunodeficiency characterized by susceptibility to viral infections, atopic eczema, defective T-cell activation and T(h)17 cell differentiation, and impaired eosinophil homeostasis and dysregulation of IgE.
高免疫球蛋白 E 综合征的遗传病因多种多样。大约 60%至 70%的高免疫球蛋白 E 综合征患者存在 STAT3 显性突变,有一例患者报道存在 TYK2 纯合突变。在其余高免疫球蛋白 E 综合征患者中,遗传病因尚未确定。
我们旨在鉴定常染色体隐性遗传高免疫球蛋白 E 综合征中发生突变或缺失的基因。
我们对 9 例常染色体隐性遗传高免疫球蛋白 E 综合征患者进行全基因组单核苷酸多态性分析,以定位拷贝数变异和纯合子单倍型。对来自另外 7 个家系的 12 例患者进行纯合子作图。对候选基因进行基因组和 cDNA 测序分析,以鉴定总共 27 例常染色体隐性遗传高免疫球蛋白 E 综合征患者的致病等位基因。
在 5 例患者中鉴定出 9 号染色体短臂亚端粒的双等位基因微缺失。在所有 5 例患者中,缺失的间隔均涉及胞质分裂因子 8(DOCK8),该基因编码一种参与调节肌动蛋白细胞骨架的蛋白。在无大片段缺失的患者中进行测序,发现来自 9 个无关家系的 16 例患者存在 DOCK8 基因的不同纯合突变,导致过早终止、移码、剪接位点破坏以及单外显子缺失和微缺失。DOCK8 缺乏与 CD4+和 CD8+T 细胞激活受损相关。
DOCK8 的常染色体隐性突变是许多(但并非全部)常染色体隐性遗传高免疫球蛋白 E 综合征的原因。DOCK8 破坏与严重的细胞免疫缺陷相关,其特征为易发生病毒感染、特应性皮炎、T 细胞激活和 T(h)17 细胞分化缺陷以及嗜酸性粒细胞稳态受损和 IgE 失调。