Department of Medical Genetics, Cambridge Institute for Medical Research, University of Cambridge, Addenbrookes Hospital, Hills Road, Cambridge, UK.
Brain. 2010 Jan;133(Pt 1):93-104. doi: 10.1093/brain/awp292. Epub 2009 Dec 9.
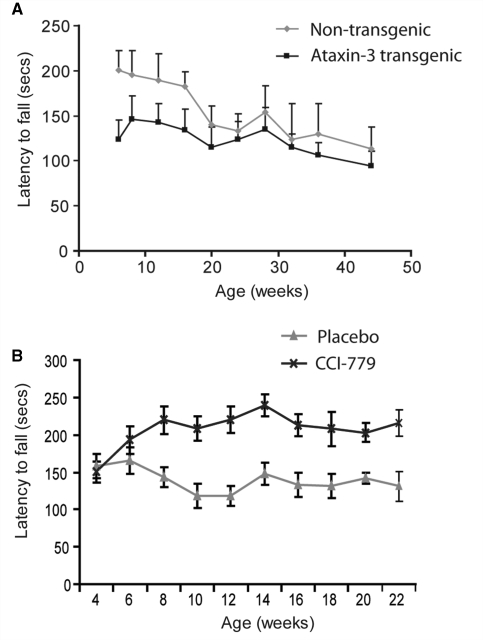
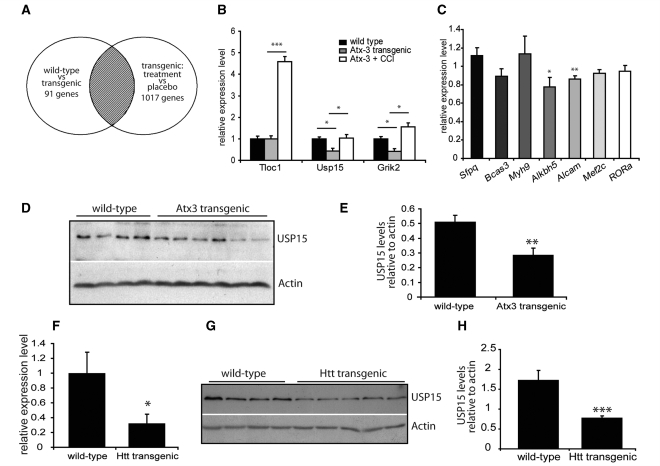
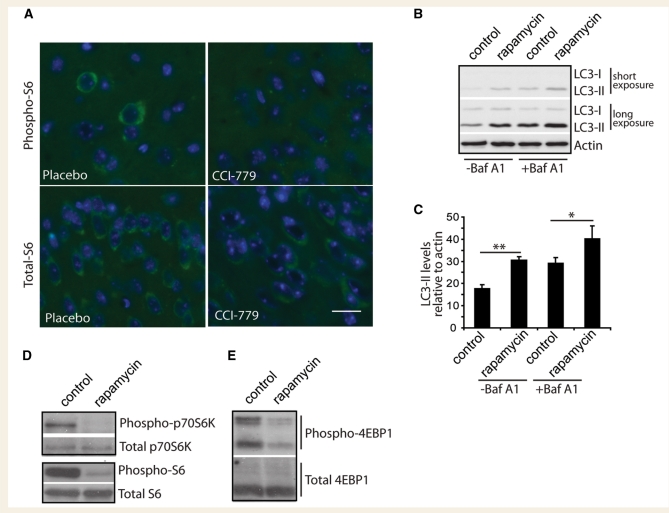
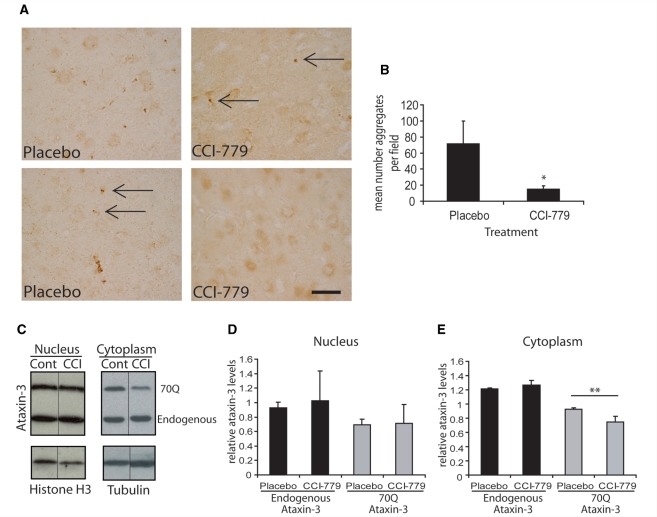
Spinocerebellar ataxia type 3 is a neurodegenerative disorder caused by the expansion of the polyglutamine repeat region within the ataxin-3 protein. The mutant protein forms intracellular aggregates in the brain. However, the cellular mechanisms causing toxicity are still poorly understood and there are currently no effective treatments. In this study we show that administration of a rapamycin ester (cell cycle inhibitor-779, temsirolimus) improves motor performance in a transgenic mouse model of spinocerebellar ataxia type 3. Temsirolimus inhibits mammalian target of rapamycin and hence upregulates protein degradation by autophagy. Temsirolimus reduces the number of aggregates seen in the brains of transgenic mice and decreases levels of cytosolic soluble mutant ataxin-3, while endogenous wild-type protein levels remain unaffected. Temsirolimus is designed for long-term use in patients and therefore represents a possible therapeutic strategy for the treatment of spinocerebellar ataxia type 3. Using this disease model and treatment paradigm, we employed a microarray approach to investigate transcriptional changes that might be important in the pathogenesis of spinocerebellar ataxia type 3. This identified ubiquitin specific peptidase-15, which showed expression changes at both the messenger ribonucleic acid and protein level. Ubiquitin specific peptidase-15 levels were also changed in mice expressing another mutant polyglutamine protein, huntingtin. In total we identified 16 transcripts that were decreased in transgenic ataxin-3 mice that were normalized following temsirolimus treatment. In this mouse model with relatively mild disease progression, the number of transcripts changed was low and the magnitude of these changes was small. However, the importance of these transcriptional alterations in the pathogenesis of spinocerebellar ataxia type 3 remains unclear.
脊髓小脑共济失调 3 型是一种神经退行性疾病,由 ataxin-3 蛋白内的多聚谷氨酰胺重复区域扩展引起。突变蛋白在大脑中形成细胞内聚集体。然而,导致毒性的细胞机制仍知之甚少,目前尚无有效的治疗方法。在这项研究中,我们表明,施用雷帕霉素酯(细胞周期抑制剂-779,替西罗莫司)可改善脊髓小脑共济失调 3 型转基因小鼠模型的运动表现。替西罗莫司抑制哺乳动物雷帕霉素靶蛋白,从而通过自噬上调蛋白降解。替西罗莫司减少了转基因小鼠大脑中聚集体的数量,并降低了细胞质可溶性突变型 ataxin-3 的水平,而内源性野生型蛋白水平保持不变。替西罗莫司专为患者的长期使用而设计,因此代表了治疗脊髓小脑共济失调 3 型的一种可能的治疗策略。使用这种疾病模型和治疗方案,我们采用了微阵列方法来研究可能对脊髓小脑共济失调 3 型发病机制重要的转录变化。这确定了泛素特异性肽酶-15,其在信使核糖核酸和蛋白质水平上均显示出表达变化。表达另一种突变型多聚谷氨酰胺蛋白亨廷顿蛋白的小鼠中,泛素特异性肽酶-15 的水平也发生了变化。我们总共鉴定了 16 种在转基因 ataxin-3 小鼠中表达降低且在替西罗莫司治疗后恢复正常的转录本。在这种疾病进展相对较轻的小鼠模型中,变化的转录本数量较少,这些变化的幅度也较小。然而,这些转录变化在脊髓小脑共济失调 3 型发病机制中的重要性尚不清楚。