Division of Medical Oncology, University of Colorado at Denver and Health Sciences Center, Aurora, Colorado, USA.
J Thorac Oncol. 2009 Dec;4(12):1455-65. doi: 10.1097/JTO.0b013e3181bc9419.
Loss of E-cadherin confers a poor prognosis in lung cancer patients and is associated with in vitro resistance to endothelial growth factor receptor inhibitors. Zinc finger E box-binding homeobox (ZEB)-1, the predominant transcriptional suppressor of E-cadherin in lung tumor lines, recruits histone deacetylases (HDACs) as co-repressors.
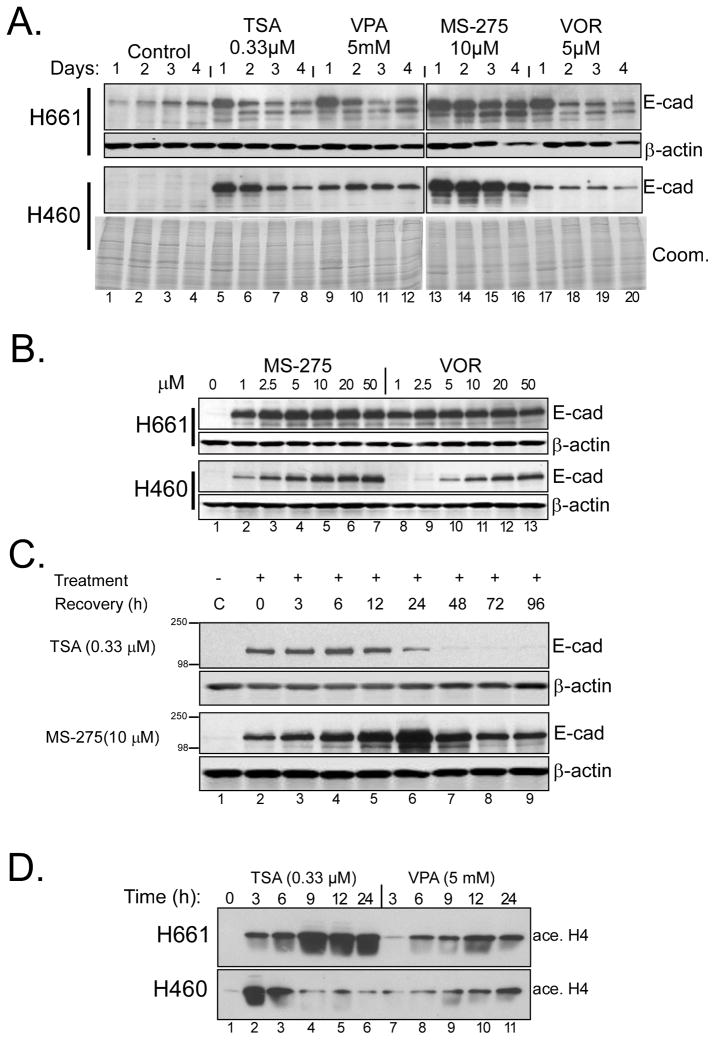
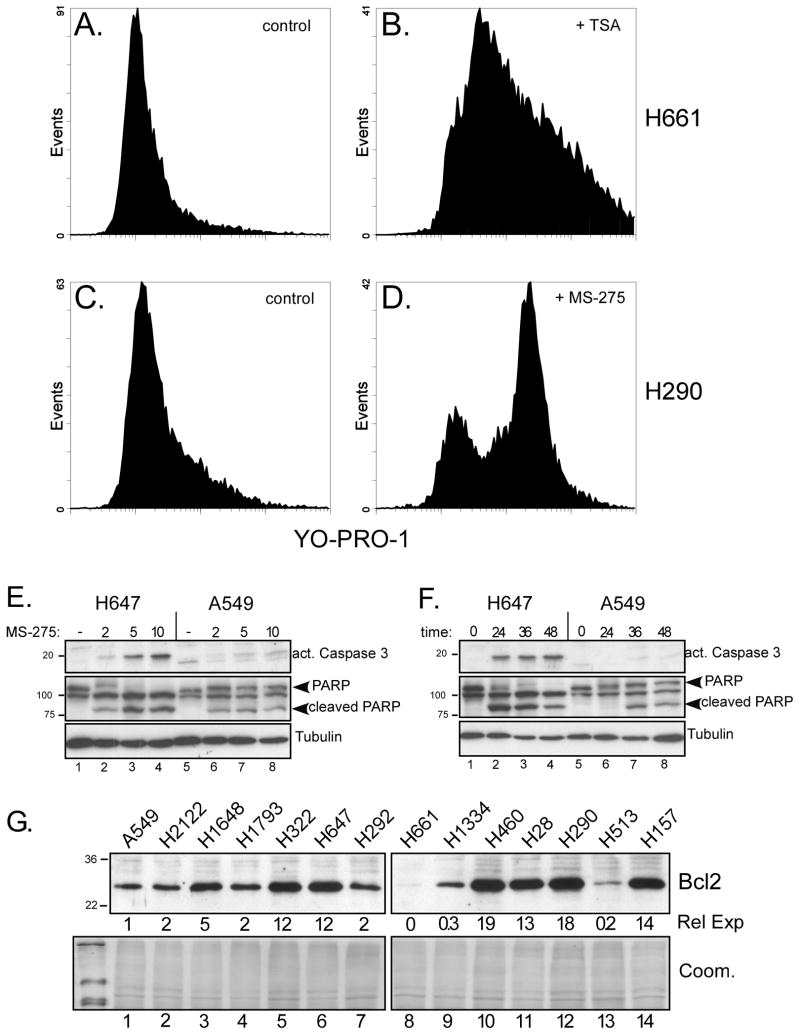
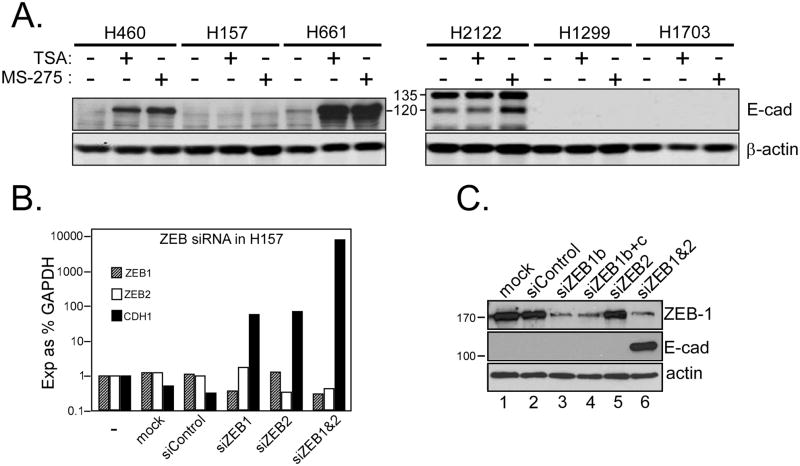
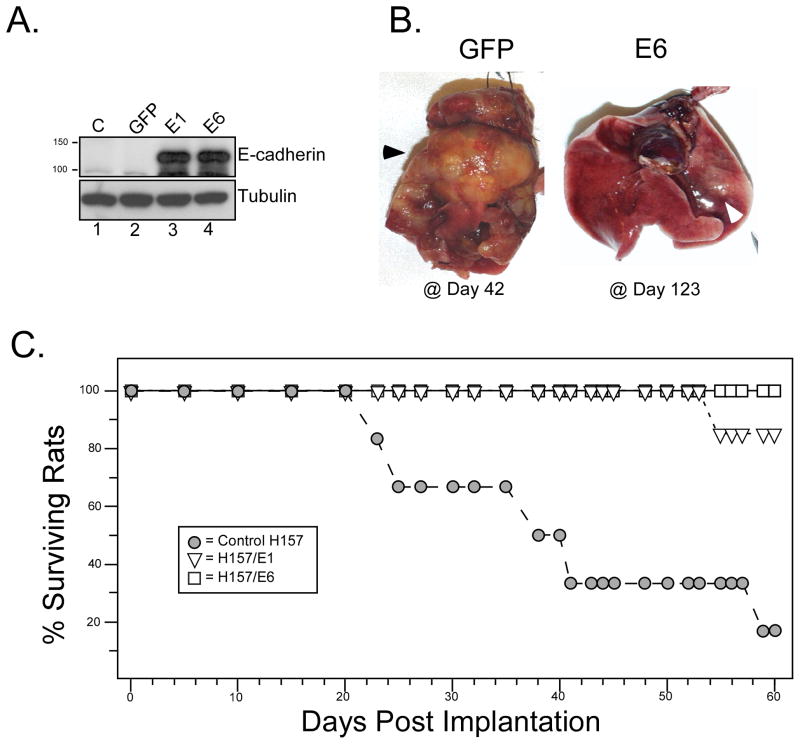
NSCLC cell lines were treated with HDAC inhibitors and analyzed for E-cadherin induction, growth inhibition and apoptosis. National Cancer Institute-H157 cells expressing ectopic E-cadherin were tested for tumorigenicity in murine xenografts.
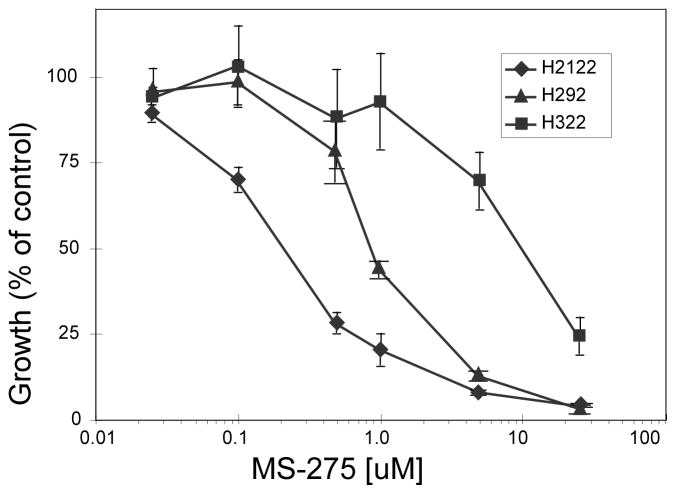
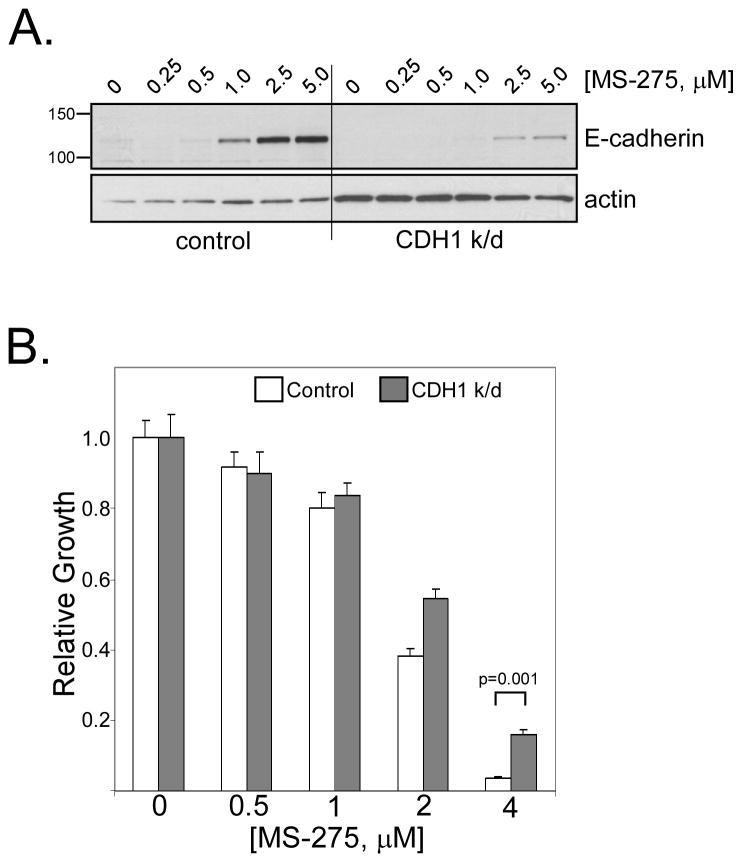
We found that treatment with MS-275, compared to vorinostat (SAHA), valproic acid or trichostatin A, was most effective in E-cadherin up-regulation and persistence in non-small cell lung cancers. As with other tumor types and HDAC inhibitors, MS-275 inhibited growth and induced apoptosis. Importantly, blocking E-cadherin induction by short hairpin RNA resulted in less inhibition by MS-275, implicating the epithelial to mesenchymal phenotype process as a contributing factor. In contrast to H460 and H661, H157 cells were resistant to E-cadherin up-regulation by HDAC inhibitors. However, E-cadherin was restored, in a synergistic manner, by combined knockdown of ZEB-1 and ZEB-2. In addition, H157 cells stably transfected with E-cadherin were markedly attenuated in their tumor forming ability. Lastly, combining MS-275 with the microtubule stabilizing agent, paclitaxel, or 17-(allylamino)-17-demethoxygeldanamycin, a heat shock protein 90 inhibitor, resulted in synergistic growth inhibition. Since MS-275 has no reported activity against HDAC6, which regulates both microtubule and heat shock protein 90 functions, other mechanisms of synergy are anticipated.
These results support the role of ZEB proteins and HDAC inhibitors in the pathogenesis and treatment of lung cancer.
E-钙黏蛋白的丢失预示着肺癌患者预后不良,并与体外对内皮生长因子受体抑制剂的耐药性相关。锌指 E 盒结合同源盒(ZEB)-1 是肺肿瘤系中 E-钙黏蛋白的主要转录抑制子,它招募组蛋白去乙酰化酶(HDACs)作为共抑制子。
用 HDAC 抑制剂处理 NSCLC 细胞系,分析 E-钙黏蛋白诱导、生长抑制和凋亡。在鼠异种移植中测试表达异位 E-钙黏蛋白的国家癌症研究所 H157 细胞的致瘤性。
与伏立诺他(SAHA)、丙戊酸或曲古抑菌素 A 相比,MS-275 处理在非小细胞肺癌中更有效地上调和维持 E-钙黏蛋白。与其他肿瘤类型和 HDAC 抑制剂一样,MS-275 抑制生长并诱导凋亡。重要的是,短发夹 RNA 阻断 E-钙黏蛋白诱导导致 MS-275 的抑制作用减弱,这表明上皮-间充质表型过程是一个促成因素。与 H460 和 H661 相反,H157 细胞对 HDAC 抑制剂上调 E-钙黏蛋白具有抗性。然而,通过同时敲低 ZEB-1 和 ZEB-2,以协同方式恢复 E-钙黏蛋白。此外,稳定转染 E-钙黏蛋白的 H157 细胞在其肿瘤形成能力方面显著减弱。最后,将 MS-275 与微管稳定剂紫杉醇或热休克蛋白 90 抑制剂 17-(烯丙基氨基)-17-去甲氧基格尔德霉素联合使用,导致协同生长抑制。由于 MS-275 对调节微管和热休克蛋白 90 功能的 HDAC6 没有报道的活性,因此预计会有其他协同作用机制。
这些结果支持 ZEB 蛋白和 HDAC 抑制剂在肺癌发病机制和治疗中的作用。