Department of Computer Science and Institute for Complex Additive Systems Analysis, New Mexico Tech, Socorro, New Mexico, United States of America.
PLoS One. 2009 Dec 11;4(12):e8250. doi: 10.1371/journal.pone.0008250.
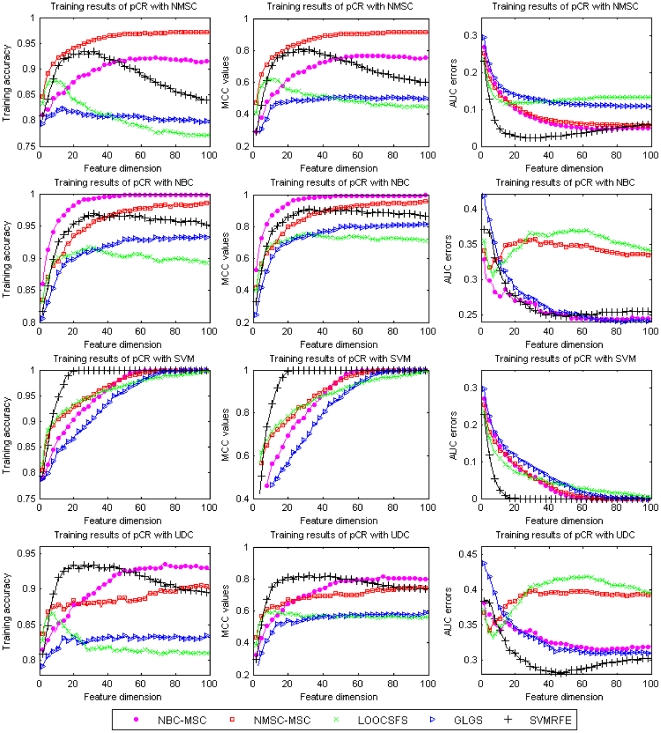
Microarray data has a high dimension of variables but available datasets usually have only a small number of samples, thereby making the study of such datasets interesting and challenging. In the task of analyzing microarray data for the purpose of, e.g., predicting gene-disease association, feature selection is very important because it provides a way to handle the high dimensionality by exploiting information redundancy induced by associations among genetic markers. Judicious feature selection in microarray data analysis can result in significant reduction of cost while maintaining or improving the classification or prediction accuracy of learning machines that are employed to sort out the datasets. In this paper, we propose a gene selection method called Recursive Feature Addition (RFA), which combines supervised learning and statistical similarity measures. We compare our method with the following gene selection methods: Support Vector Machine Recursive Feature Elimination (SVMRFE), Leave-One-Out Calculation Sequential Forward Selection (LOOCSFS), Gradient based Leave-one-out Gene Selection (GLGS). To evaluate the performance of these gene selection methods, we employ several popular learning classifiers on the MicroArray Quality Control phase II on predictive modeling (MAQC-II) breast cancer dataset and the MAQC-II multiple myeloma dataset. Experimental results show that gene selection is strictly paired with learning classifier. Overall, our approach outperforms other compared methods. The biological functional analysis based on the MAQC-II breast cancer dataset convinced us to apply our method for phenotype prediction. Additionally, learning classifiers also play important roles in the classification of microarray data and our experimental results indicate that the Nearest Mean Scale Classifier (NMSC) is a good choice due to its prediction reliability and its stability across the three performance measurements: Testing accuracy, MCC values, and AUC errors.
微阵列数据具有很高的变量维度,但可用的数据集通常只有少量的样本,因此研究此类数据集非常有趣且具有挑战性。在分析微阵列数据的任务中,例如预测基因-疾病关联,特征选择非常重要,因为它提供了一种利用遗传标记之间的关联所产生的信息冗余来处理高维数据的方法。在微阵列数据分析中进行明智的特征选择可以显著降低成本,同时保持或提高用于整理数据集的学习机器的分类或预测准确性。在本文中,我们提出了一种称为递归特征添加(RFA)的基因选择方法,该方法结合了监督学习和统计相似性度量。我们将我们的方法与以下基因选择方法进行了比较:支持向量机递归特征消除(SVMRFE)、留一计算顺序前向选择(LOOCSFS)、基于梯度的留一基因选择(GLGS)。为了评估这些基因选择方法的性能,我们在 MicroArray Quality Control phase II on predictive modeling(MAQC-II)乳腺癌数据集和 MAQC-II 多发性骨髓瘤数据集上使用了几种流行的学习分类器。实验结果表明,基因选择与学习分类器严格配对。总的来说,我们的方法优于其他比较方法。基于 MAQC-II 乳腺癌数据集的生物学功能分析使我们相信可以应用我们的方法进行表型预测。此外,学习分类器在微阵列数据的分类中也起着重要作用,我们的实验结果表明,由于其预测可靠性及其在三个性能测量中的稳定性,最近均值尺度分类器(NMSC)是一个不错的选择:测试准确性、MCC 值和 AUC 误差。