Tintle Nathan L, Borchers Bryce, Brown Marshall, Bekmetjev Airat
Department of Mathematics, Hope College, 27 Graves Place, Holland, Michigan 49423, USA.
BMC Proc. 2009 Dec 15;3 Suppl 7(Suppl 7):S96. doi: 10.1186/1753-6561-3-s7-s96.
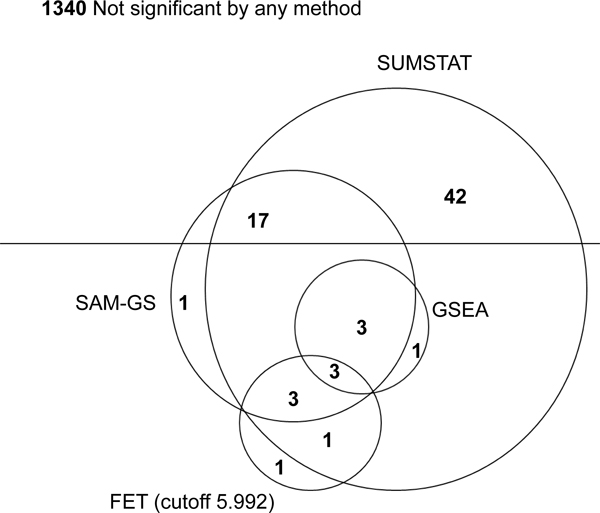
Recently, gene set analysis (GSA) has been extended from use on gene expression data to use on single-nucleotide polymorphism (SNP) data in genome-wide association studies. When GSA has been demonstrated on SNP data, two popular statistics from gene expression data analysis (gene set enrichment analysis [GSEA] and Fisher's exact test [FET]) have been used. However, GSEA and FET have shown a lack of power and robustness in the analysis of gene expression data. The purpose of this work is to investigate whether the same issues are also true for the analysis of SNP data. Ultimately, we conclude that GSEA and FET are not optimal for the analysis of SNP data when compared with the SUMSTAT method. In analysis of real SNP data from the Framingham Heart Study, we find that SUMSTAT finds many more gene sets to be significant when compared with other methods. In an analysis of simulated data, SUMSTAT demonstrates high power and better control of the type I error rate. GSA is a promising approach to the analysis of SNP data in GWAS and use of the SUMSTAT statistic instead of GSEA or FET may increase power and robustness.
最近,基因集分析(GSA)已从用于基因表达数据扩展到用于全基因组关联研究中的单核苷酸多态性(SNP)数据。当在SNP数据上进行GSA验证时,人们使用了来自基因表达数据分析的两种常用统计方法(基因集富集分析[GSEA]和Fisher精确检验[FET])。然而,GSEA和FET在基因表达数据分析中已显示出缺乏效力和稳健性。这项工作的目的是研究在SNP数据分析中是否也存在同样的问题。最终,我们得出结论,与SUMSTAT方法相比,GSEA和FET在SNP数据分析中并非最优。在对弗雷明汉心脏研究的真实SNP数据进行分析时,我们发现与其他方法相比,SUMSTAT发现的显著基因集更多。在对模拟数据的分析中,SUMSTAT显示出高效力和对I型错误率的更好控制。GSA是全基因组关联研究中SNP数据分析的一种有前景的方法,使用SUMSTAT统计量而非GSEA或FET可能会提高效力和稳健性。