Rosta Edina, Buchete Nicolae-Viorel, Hummer Gerhard
Laboratory of Chemical Physics, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892-0520, U.S.A., and School of Physics, University College Dublin, Belfield, Dublin 4, Ireland.
J Chem Theory Comput. 2009;5(5):1393-1399. doi: 10.1021/ct800557h.
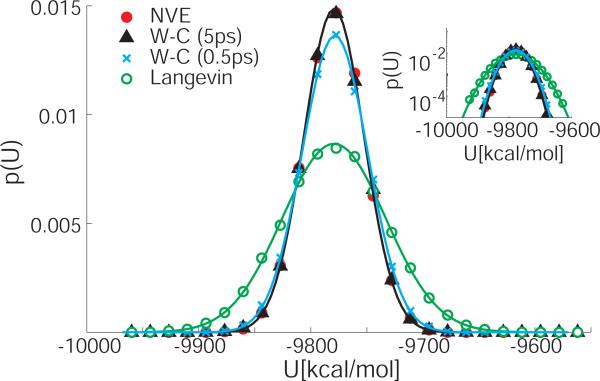
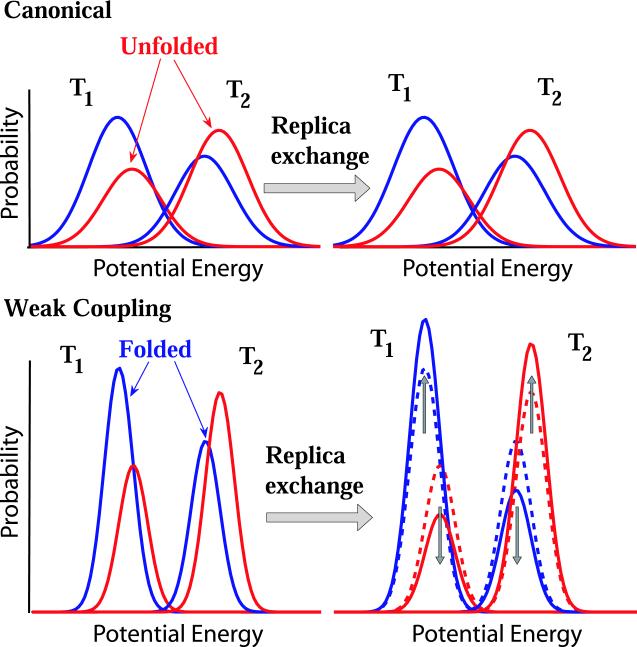
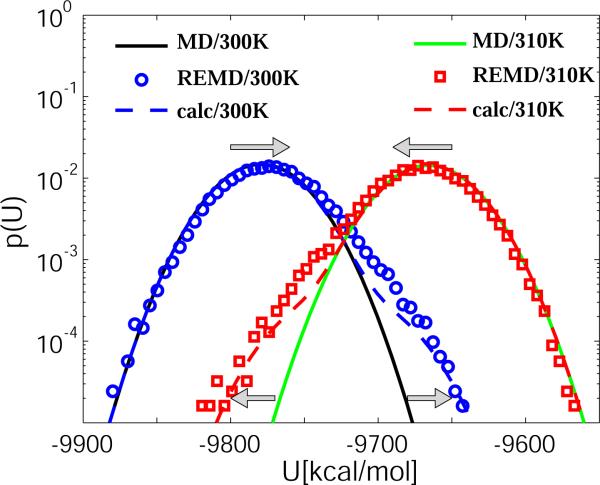
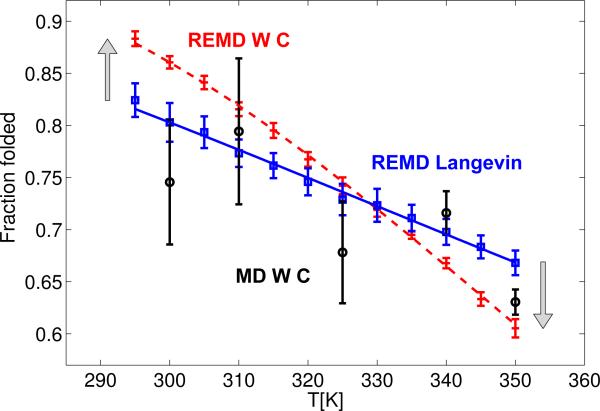
We explore the effects of thermostats in replica exchange molecular dynamics (REMD) simulations. For thermostats that do not produce a canonical ensemble, REMD simulations are found to distort the configuration-space distributions. For bulk water, we find small deviations of the average potential energies, the buildup of tails in the potential energy distributions, and artificial correlations between the energies at different temperatures. If a solute is present, as in protein folding simulations, its conformational equilibrium can be altered. In REMD simulations of a helix-forming peptide with a weak-coupling (Berendsen) thermostat, we find that the folded state is overpopulated by about 10% at low temperatures, and underpopulated at high temperatures. As a consequence, the enthalpy of folding deviates by almost 3 kcal/mol from the correct value. The reason for this population shift is that non-canonical ensembles with narrowed potential energy fluctuations artificially bias toward replica exchanges between low-energy folded structures at the high temperature and high-energy unfolded structures at the low temperature. We conclude that REMD simulations should only be performed in conjunction with thermostats that produce a canonical ensemble.
我们探讨了恒温器在副本交换分子动力学(REMD)模拟中的作用。对于那些不能产生正则系综的恒温器,我们发现REMD模拟会扭曲构型空间分布。对于体相水,我们发现平均势能存在小偏差、势能分布出现尾部积累以及不同温度下能量之间存在人为相关性。如果存在溶质,如在蛋白质折叠模拟中,其构象平衡可能会改变。在使用弱耦合(Berendsen)恒温器对形成螺旋的肽进行REMD模拟时,我们发现在低温下折叠态的占比高出约10%,而在高温下则占比不足。因此,折叠焓与正确值相差近3千卡/摩尔。这种占比变化的原因是,势能涨落变窄的非正则系综会人为地偏向于高温下低能量折叠结构与低温下高能量未折叠结构之间的副本交换。我们得出结论,REMD模拟应该仅与能产生正则系综的恒温器一起进行。