Kahn Crystal L, Mozes Shay, Raphael Benjamin J
Algorithms Mol Biol. 2010 Jan 4;5(1):11. doi: 10.1186/1748-7188-5-11.
Segmental duplications, or low-copy repeats, are common in mammalian genomes. In the human genome, most segmental duplications are mosaics comprised of multiple duplicated fragments. This complex genomic organization complicates analysis of the evolutionary history of these sequences. One model proposed to explain this mosaic patterns is a model of repeated aggregation and subsequent duplication of genomic sequences.
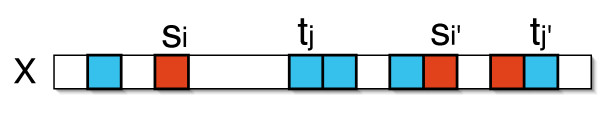
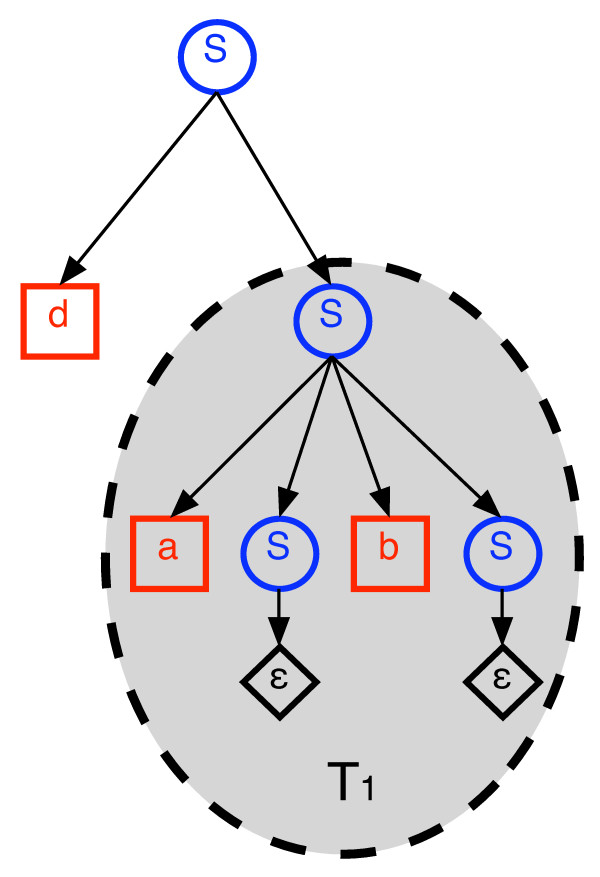
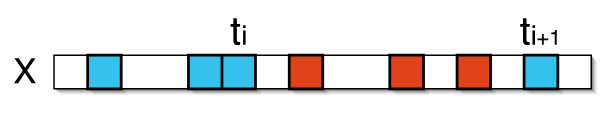
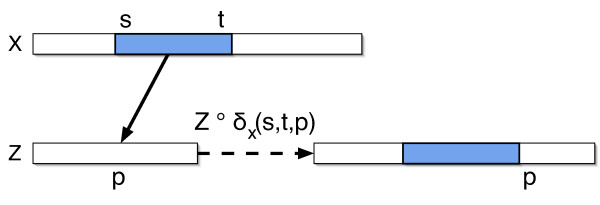
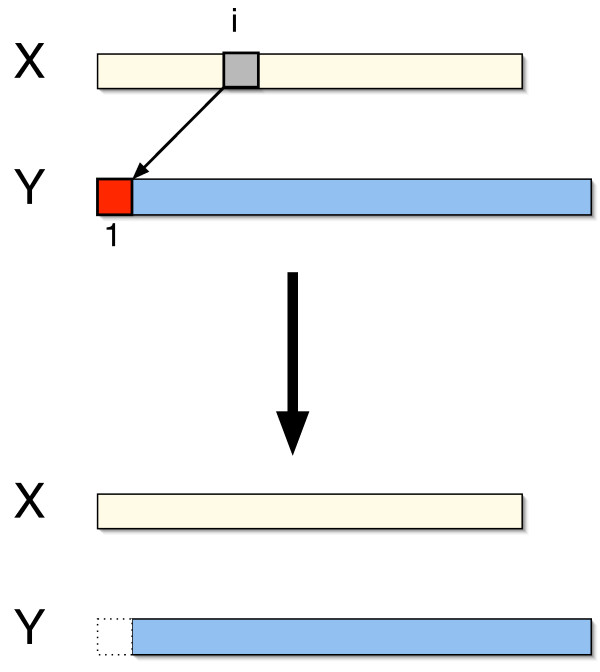
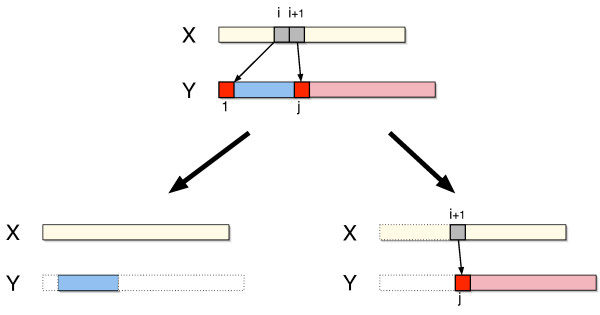
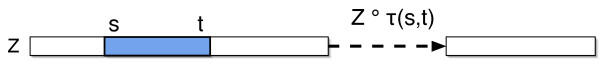
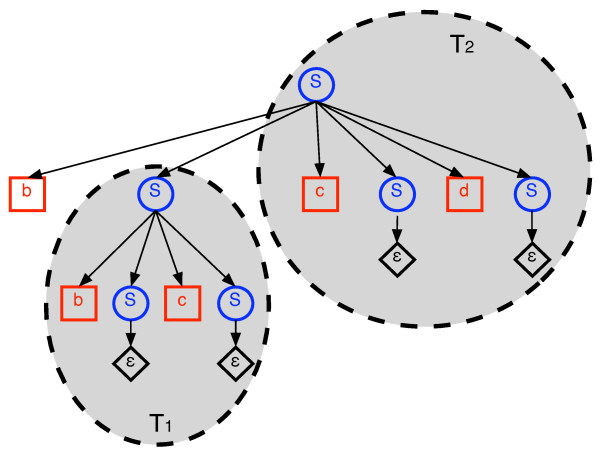
We describe a polynomial-time exact algorithm to compute duplication distance, a genomic distance defined as the most parsimonious way to build a target string by repeatedly copying substrings of a fixed source string. This distance models the process of repeated aggregation and duplication. We also describe extensions of this distance to include certain types of substring deletions and inversions. Finally, we provide a description of a sequence of duplication events as a context-free grammar (CFG).
These new genomic distances will permit more biologically realistic analyses of segmental duplications in genomes.
片段重复,即低拷贝重复,在哺乳动物基因组中很常见。在人类基因组中,大多数片段重复是由多个重复片段组成的镶嵌体。这种复杂的基因组组织使这些序列的进化历史分析变得复杂。为解释这种镶嵌模式而提出的一种模型是基因组序列重复聚集和随后重复的模型。
我们描述了一种多项式时间精确算法来计算重复距离,这是一种基因组距离,定义为通过重复复制固定源字符串的子串来构建目标字符串的最简约方式。这个距离模拟了重复聚集和重复的过程。我们还描述了这个距离的扩展,以包括某些类型的子串删除和倒置。最后,我们将重复事件序列描述为上下文无关语法(CFG)。
这些新的基因组距离将允许对基因组中的片段重复进行更符合生物学实际的分析。