USDA, ARS, ANRI, Bovine Functional Genomics Laboratory, Beltsville, Maryland 20705, USA.
BMC Genomics. 2009 Dec 1;10:571. doi: 10.1186/1471-2164-10-571.
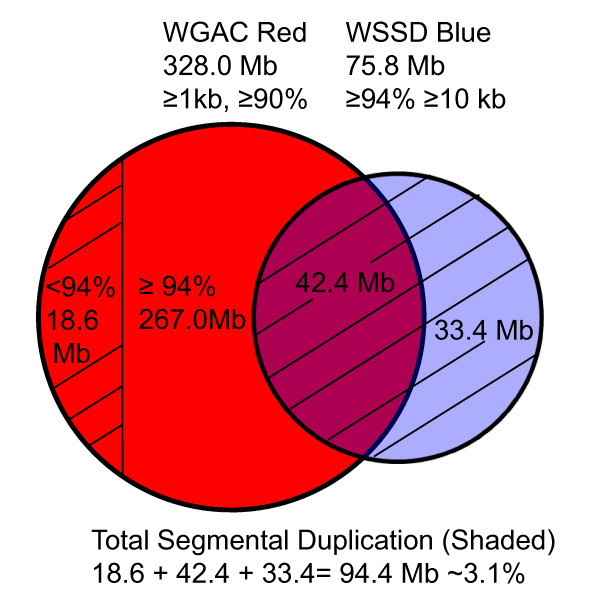
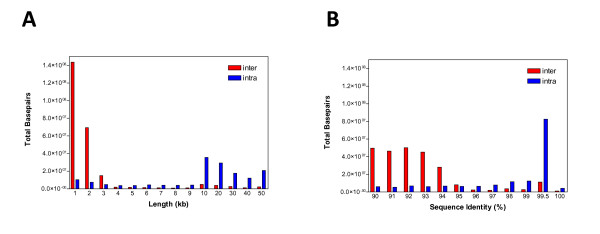
Duplicated sequences are an important source of gene innovation and structural variation within mammalian genomes. We performed the first systematic and genome-wide analysis of segmental duplications in the modern domesticated cattle (Bos taurus). Using two distinct computational analyses, we estimated that 3.1% (94.4 Mb) of the bovine genome consists of recently duplicated sequences (>or= 1 kb in length, >or= 90% sequence identity). Similar to other mammalian draft assemblies, almost half (47% of 94.4 Mb) of these sequences have not been assigned to cattle chromosomes.
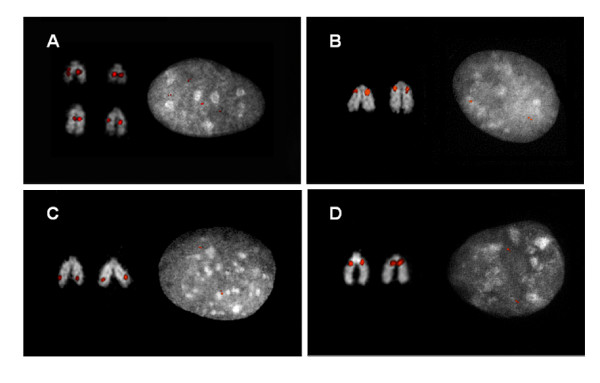
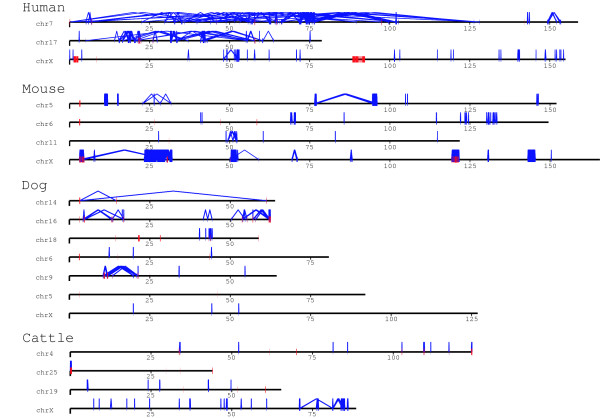
In this study, we provide the first experimental validation large duplications and briefly compared their distribution on two independent bovine genome assemblies using fluorescent in situ hybridization (FISH). Our analyses suggest that the (75-90%) of segmental duplications are organized into local tandem duplication clusters. Along with rodents and carnivores, these results now confidently establish tandem duplications as the most likely mammalian archetypical organization, in contrast to humans and great ape species which show a preponderance of interspersed duplications. A cross-species survey of duplicated genes and gene families indicated that duplication, positive selection and gene conversion have shaped primates, rodents, carnivores and ruminants to different degrees for their speciation and adaptation. We identified that bovine segmental duplications corresponding to genes are significantly enriched for specific biological functions such as immunity, digestion, lactation and reproduction.
Our results suggest that in most mammalian lineages segmental duplications are organized in a tandem configuration. Segmental duplications remain problematic for genome and assembly and we highlight genic regions that require higher quality sequence characterization. This study provides insights into mammalian genome evolution and generates a valuable resource for cattle genomics research.
重复序列是哺乳动物基因组中基因创新和结构变异的重要来源。我们首次对现代家养牛(Bos taurus)中的片段重复进行了系统的全基因组分析。通过两种不同的计算分析,我们估计牛基因组的 3.1%(94.4Mb)由最近复制的序列组成(长度> = 1kb,序列同一性> = 90%)。与其他哺乳动物草图组装类似,这些序列中有近一半(94.4Mb 的 47%)尚未分配给牛染色体。
在这项研究中,我们首次提供了大重复的实验验证,并使用荧光原位杂交(FISH)简要比较了它们在两个独立的牛基因组组装上的分布。我们的分析表明,(75-90%)的片段重复被组织成局部串联重复簇。与啮齿动物和食肉动物一样,这些结果现在可以确定串联重复是最有可能的哺乳动物原型组织,而与人类和大型猿类物种形成鲜明对比,这些物种表现出散布重复的优势。对重复基因和基因家族的跨物种调查表明,复制、正选择和基因转换在不同程度上塑造了灵长类动物、啮齿动物、食肉动物和反刍动物,以适应其物种形成和适应。我们确定与基因对应的牛片段重复明显富集了特定的生物学功能,如免疫、消化、泌乳和繁殖。
我们的结果表明,在大多数哺乳动物谱系中,片段重复以串联形式组织。片段重复仍然是基因组和组装的问题,我们强调了需要更高质量序列特征的基因区域。这项研究为哺乳动物基因组进化提供了新的见解,并为牛基因组学研究提供了有价值的资源。