University of Central Florida College of Medicine, Orlando, 32827, USA.
Biochem Pharmacol. 2010 May 15;79(10):1398-409. doi: 10.1016/j.bcp.2010.01.001. Epub 2010 Jan 11.
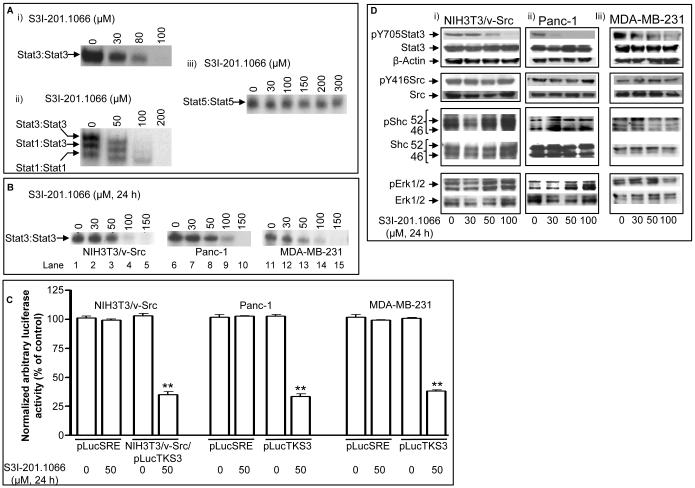
The molecular modeling of the phosphotyrosine (pTyr)-SH2 domain interaction in the Stat3:Stat3 dimerization, combined with in silico structural analysis of the Stat3 dimerization disruptor, S3I-201, has furnished a diverse set of analogs. We present evidence from in vitro biochemical and biophysical studies that the structural analog, S3I-201.1066 directly interacts with Stat3 or the SH2 domain, with an affinity (K(D)) of 2.74microM, and disrupts the binding of Stat3 to the cognate pTyr-peptide, GpYLPQTV-NH(2), with an IC(50) of 23microM. Moreover, S3I-201.1066 selectively blocks the association of Stat3 with the epidermal growth factor receptor (EGFR), and inhibits Stat3 tyrosine phosphorylation and nuclear translocation in EGF-stimulated mouse fibroblasts. In cancer cells that harbor aberrant Stat3 activity, S3I-201.1066 inhibits constitutive Stat3 DNA-binding and transcriptional activities. By contrast, S3I-201.1066 has no effect on Src activation or the EGFR-mediated activation of the Erk1/2(MAPK) pathway. S3I-201.1066 selectively suppresses the viability, survival, and malignant transformation of the human breast and pancreatic cancer lines and the v-Src-transformed mouse fibroblasts harboring persistently active Stat3. Treatment with S3I-201.1066 of malignant cells harboring aberrantly active Stat3 down-regulated the expression of c-Myc, Bcl-xL, Survivin, the matrix metalloproteinase 9, and VEGF. The in vivo administration of S3I-201.1066-induced significant antitumor response in mouse models of human breast cancer, which correlates with the inhibition of constitutively active Stat3 and the suppression of known Stat3-regulated genes. Our studies identify a novel small-molecule that binds with a high affinity to Stat3, blocks Stat3 activation and function, and thereby induces antitumor response in human breast tumor xenografts harboring persistently active Stat3.
磷酸化酪氨酸(pTyr)-SH2 结构域与 Stat3 二聚化的分子建模,结合 Stat3 二聚化破坏剂 S3I-201 的计算结构分析,提供了一组不同的类似物。我们从体外生化和生物物理研究中提供证据表明,结构类似物 S3I-201.1066 直接与 Stat3 或 SH2 结构域相互作用,亲和力(K(D))为 2.74μM,并破坏 Stat3 与同源 pTyr-肽 GpYLPQTV-NH(2)的结合,IC(50)为 23μM。此外,S3I-201.1066 选择性地阻断 Stat3 与表皮生长因子受体(EGFR)的结合,并抑制 EGF 刺激的小鼠成纤维细胞中 Stat3 的酪氨酸磷酸化和核转位。在异常 Stat3 活性的癌细胞中,S3I-201.1066 抑制组成性 Stat3 DNA 结合和转录活性。相比之下,S3I-201.1066 对 Src 激活或 EGFR 介导的 Erk1/2(MAPK)途径的激活没有影响。S3I-201.1066 选择性地抑制人乳腺癌和胰腺癌系以及持续激活 Stat3 的 v-Src 转化的小鼠成纤维细胞的活力、存活和恶性转化。用 S3I-201.1066 处理异常激活 Stat3 的恶性细胞下调 c-Myc、Bcl-xL、Survivin、基质金属蛋白酶 9 和 VEGF 的表达。S3I-201.1066 在携带人乳腺癌的小鼠模型中的体内给药诱导了显著的抗肿瘤反应,这与持续激活的 Stat3 的抑制和已知的 Stat3 调节基因的抑制相关。我们的研究鉴定了一种新型小分子,它与 Stat3 高亲和力结合,阻断 Stat3 激活和功能,从而诱导携带持续激活 Stat3 的人乳腺癌肿瘤异种移植物的抗肿瘤反应。