MRC Biostatistics Unit, Institute of Public Health, Forvie Site, Cambridge, CB2 0SR, UK.
BMC Genomics. 2010 Jan 14;11:30. doi: 10.1186/1471-2164-11-30.
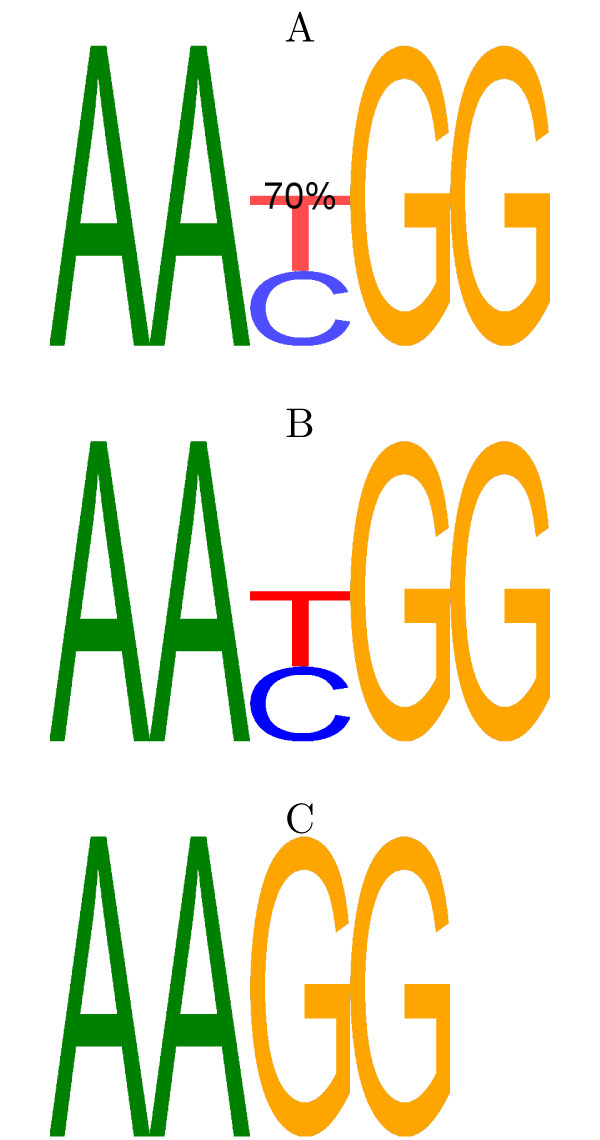
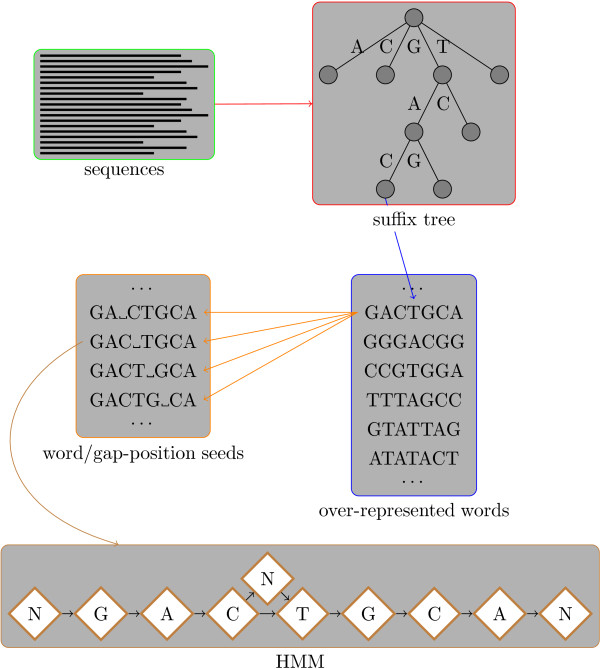
Classically, models of DNA-transcription factor binding sites (TFBSs) have been based on relatively few known instances and have treated them as sites of fixed length using position weight matrices (PWMs). Various extensions to this model have been proposed, most of which take account of dependencies between the bases in the binding sites. However, some transcription factors are known to exhibit some flexibility and bind to DNA in more than one possible physical configuration. In some cases this variation is known to affect the function of binding sites. With the increasing volume of ChIP-seq data available it is now possible to investigate models that incorporate this flexibility. Previous work on variable length models has been constrained by: a focus on specific zinc finger proteins in yeast using restrictive models; a reliance on hand-crafted models for just one transcription factor at a time; and a lack of evaluation on realistically sized data sets.
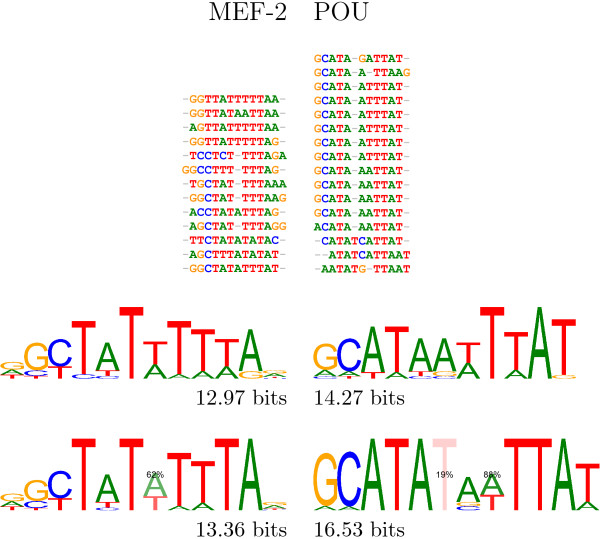
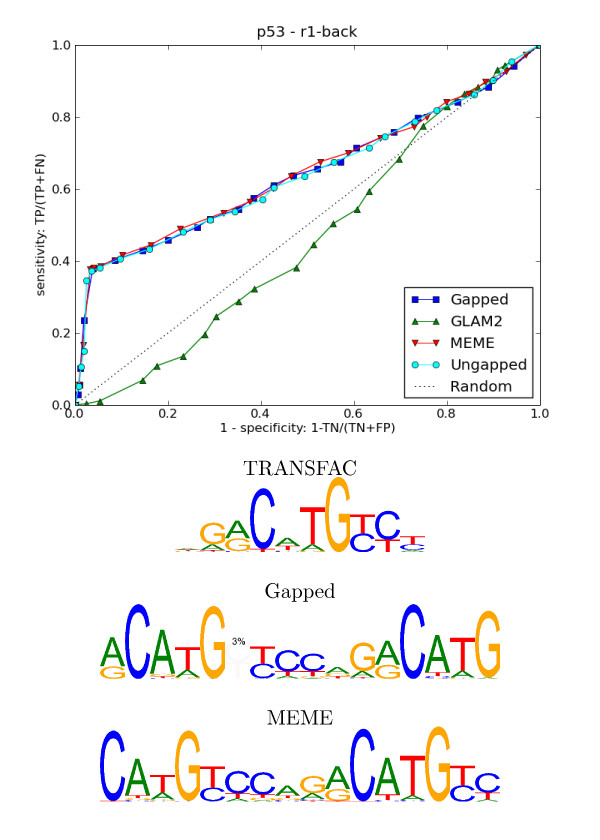
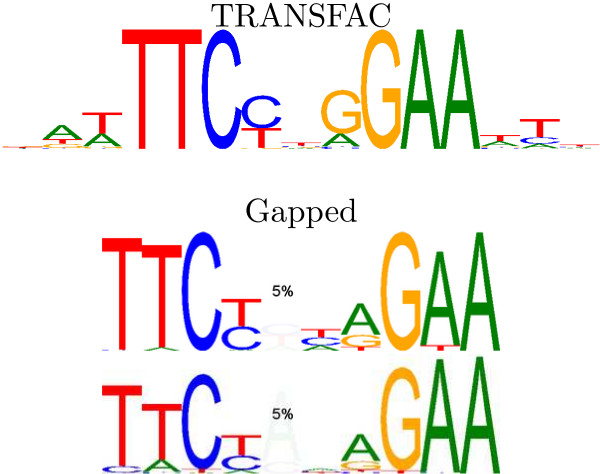
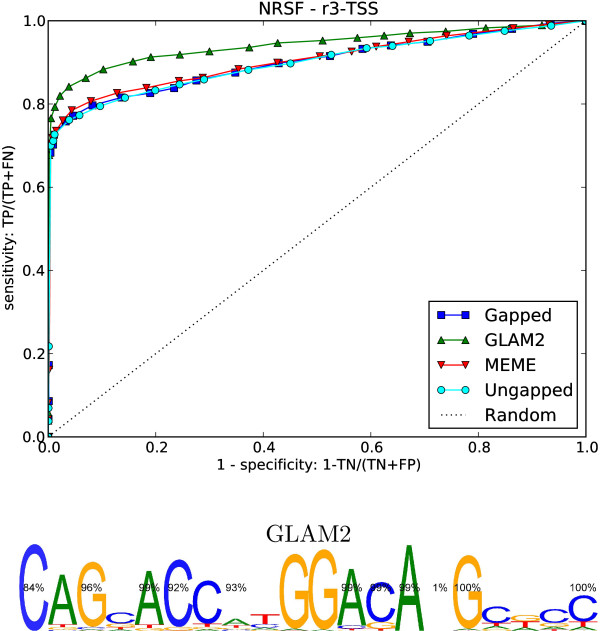
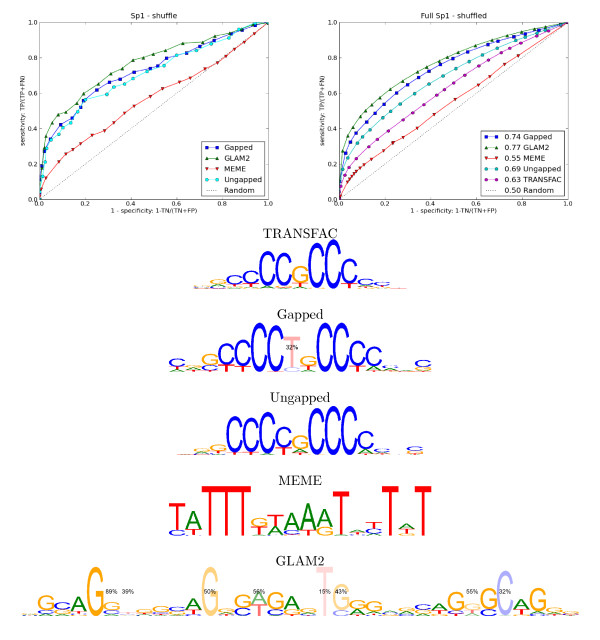
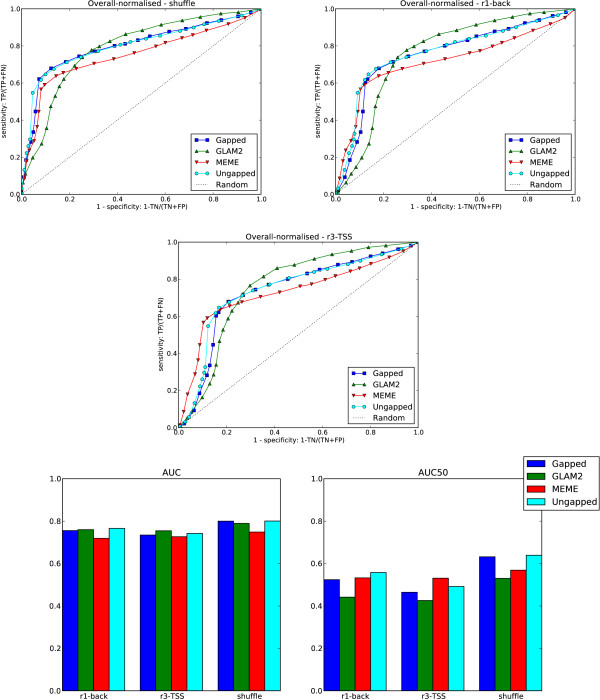
We re-analysed binding sites from the TRANSFAC database and found motivating examples where our new variable length model provides a better fit. We analysed several ChIP-seq data sets with a novel motif search algorithm and compared the results to one of the best standard PWM finders and a recently developed alternative method for finding motifs of variable structure. All the methods performed comparably in held-out cross validation tests. Known motifs of variable structure were recovered for p53, Stat5a and Stat5b. In addition our method recovered a novel generalised version of an existing PWM for Sp1 that allows for variable length binding. This motif improved classification performance.
We have presented a new gapped PWM model for variable length DNA binding sites that is not too restrictive nor over-parameterised. Our comparison with existing tools shows that on average it does not have better predictive accuracy than existing methods. However, it does provide more interpretable models of motifs of variable structure that are suitable for follow-up structural studies. To our knowledge, we are the first to apply variable length motif models to eukaryotic ChIP-seq data sets and consequently the first to show their value in this domain. The results include a novel motif for the ubiquitous transcription factor Sp1.
经典的 DNA-转录因子结合位点(TFBS)模型是基于相对较少的已知实例,并使用位置权重矩阵(PWMs)将其视为固定长度的位点。已经提出了该模型的各种扩展,其中大多数考虑了结合位点中碱基之间的依赖性。然而,已知一些转录因子表现出一定的灵活性,并以多种可能的物理构象结合 DNA。在某些情况下,这种变化已知会影响结合位点的功能。随着 ChIP-seq 数据量的增加,现在可以研究纳入这种灵活性的模型。以前关于可变长度模型的工作受到以下因素的限制:在酵母中使用限制性模型专注于特定的锌指蛋白;一次依赖于手工制作的模型来寻找一个转录因子;以及缺乏对真实大小数据集的评估。
我们重新分析了 TRANSFAC 数据库中的结合位点,并找到了一些令人信服的例子,在这些例子中,我们的新可变长度模型提供了更好的拟合。我们使用一种新的 motif 搜索算法分析了几个 ChIP-seq 数据集,并将结果与最好的标准 PWM 查找器之一和最近开发的用于寻找可变结构 motif 的替代方法进行了比较。所有方法在保留交叉验证测试中表现相当。为 p53、Stat5a 和 Stat5b 找到了可变结构的已知 motif。此外,我们的方法还为 Sp1 恢复了一个现有的 PWM 的新的通用版本,该版本允许可变长度结合。该 motif 提高了分类性能。
我们提出了一种新的可变长度 DNA 结合位点的间隙 PWM 模型,该模型既不太严格也不过度参数化。我们与现有工具的比较表明,平均而言,它的预测准确性并不优于现有方法。然而,它确实提供了更具可解释性的可变结构 motif 模型,适合后续的结构研究。据我们所知,我们是第一个将可变长度 motif 模型应用于真核生物 ChIP-seq 数据集的人,因此也是第一个在该领域展示其价值的人。结果包括一个新的普遍转录因子 Sp1 的 motif。