Tel Aviv University, Israel.
Proteins. 2010 May 1;78(6):1503-19. doi: 10.1002/prot.22668.
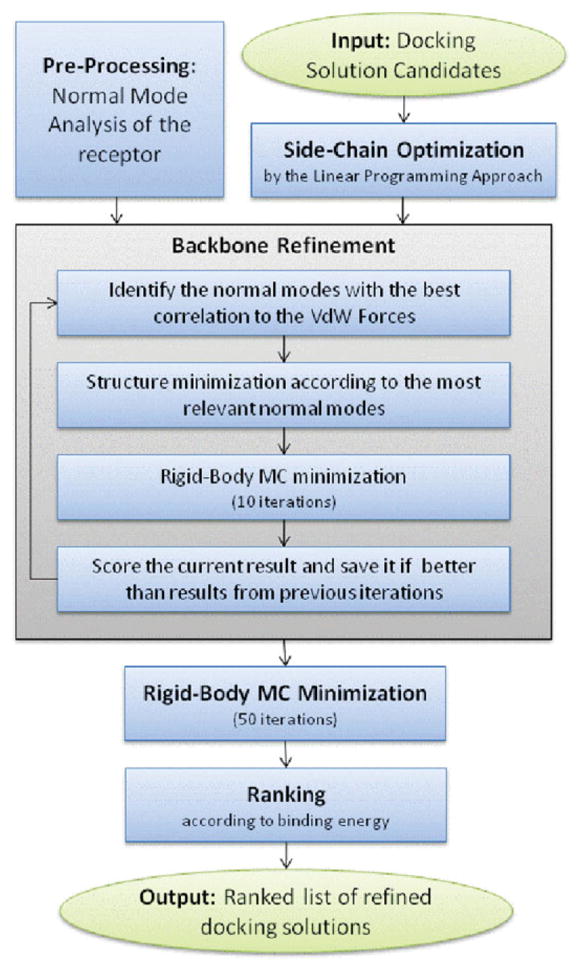
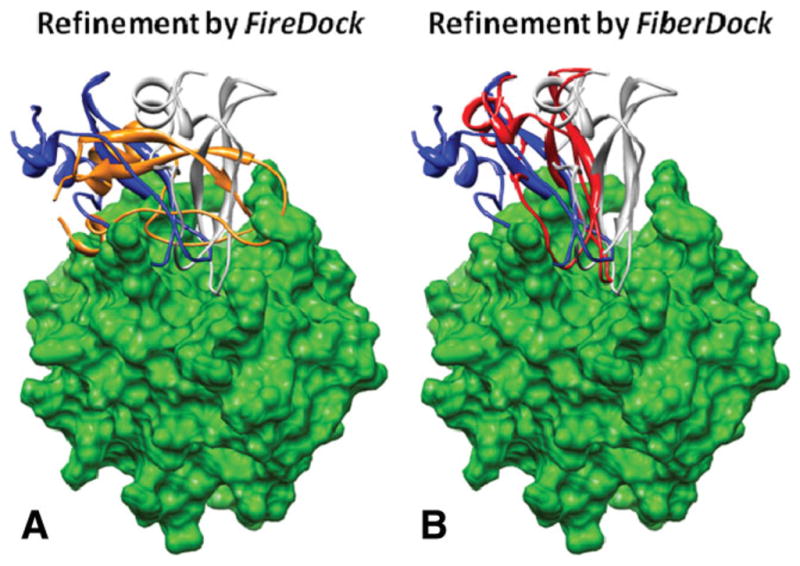
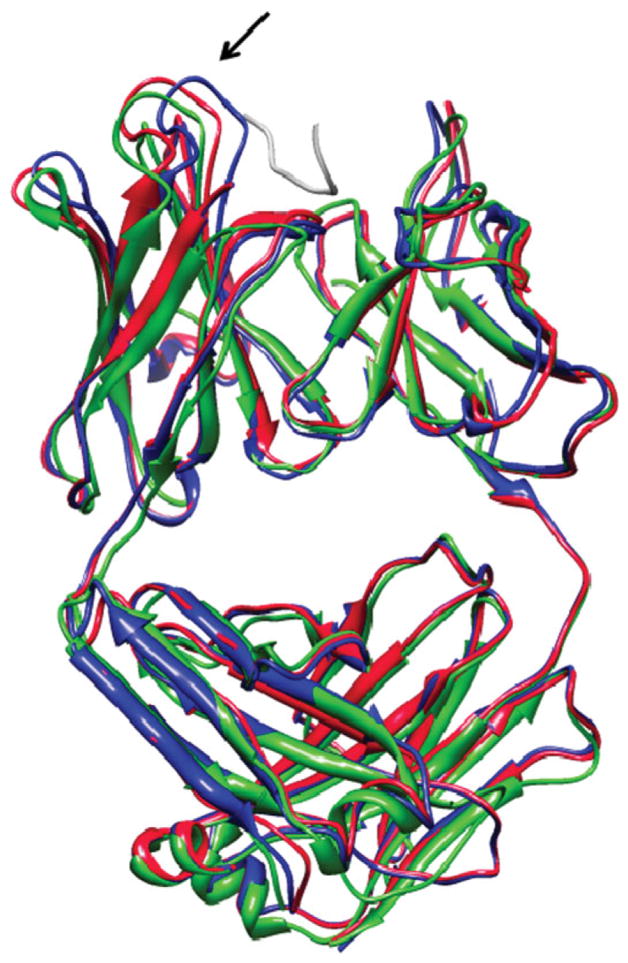
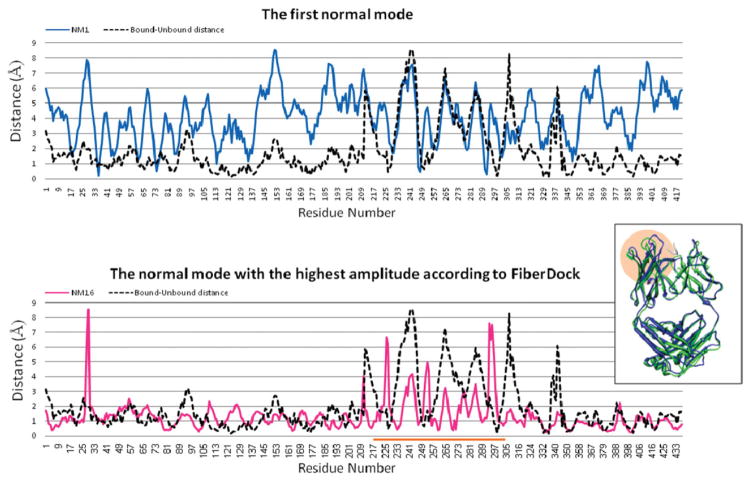
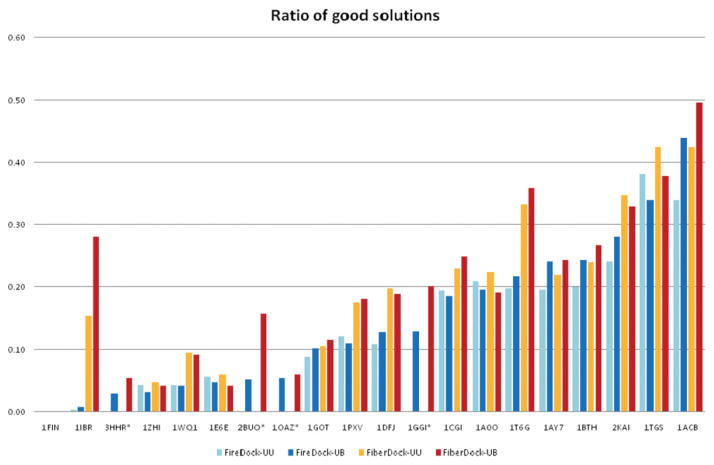
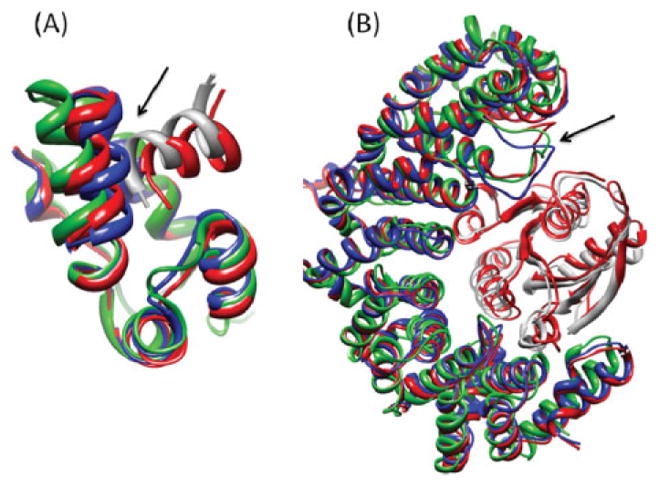
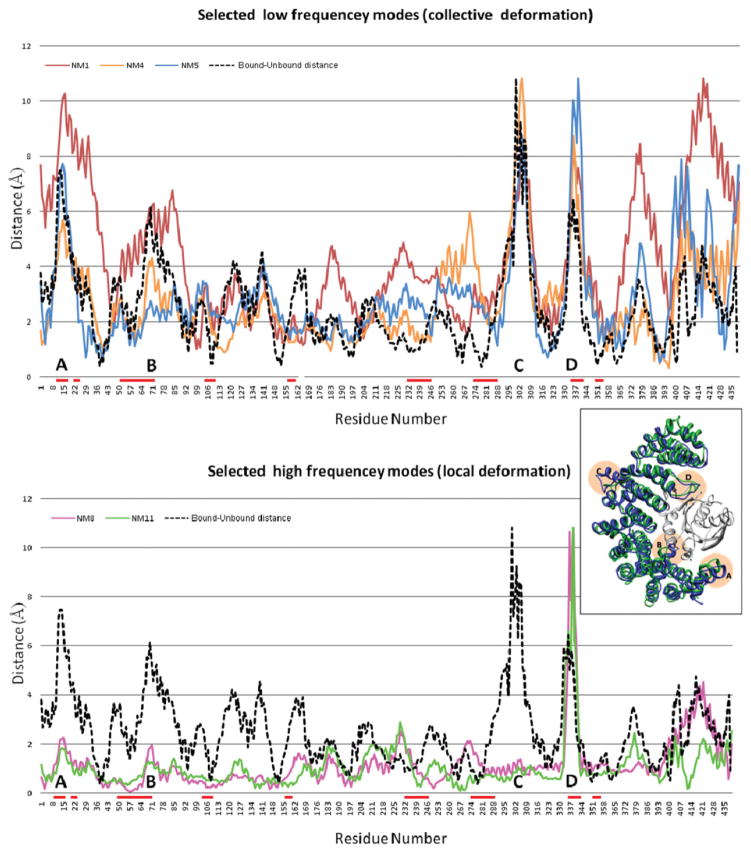
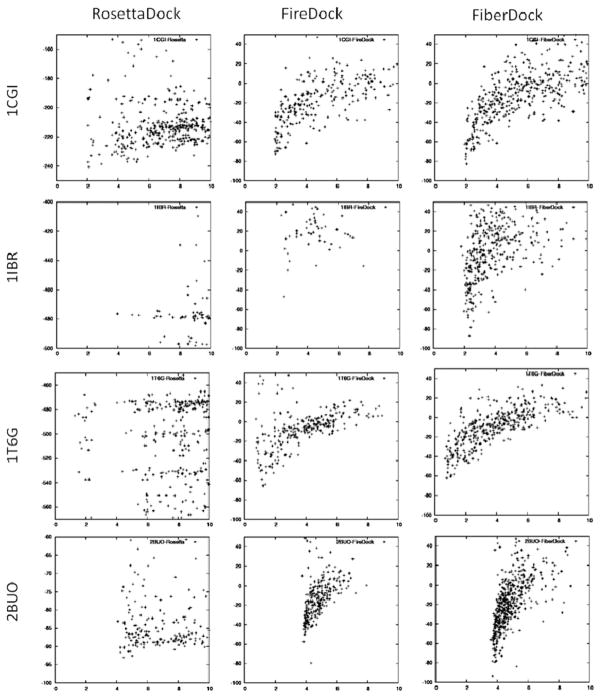
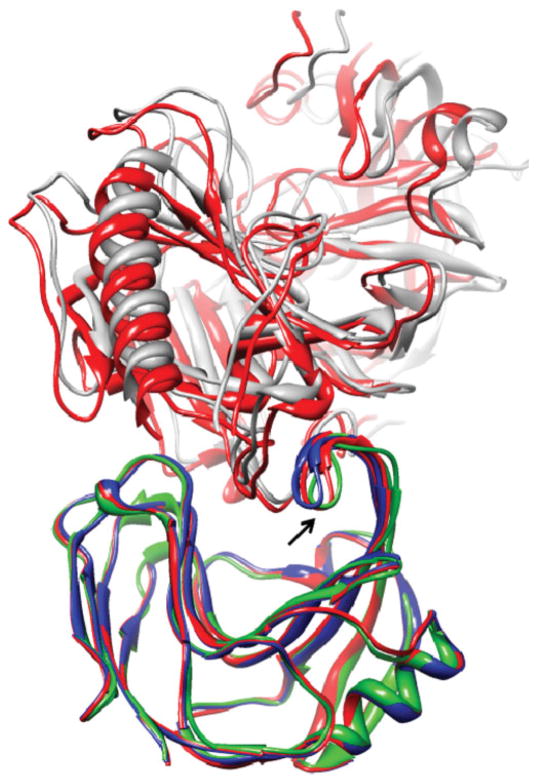
Upon binding, proteins undergo conformational changes. These changes often prevent rigid-body docking methods from predicting the 3D structure of a complex from the unbound conformations of its proteins. Handling protein backbone flexibility is a major challenge for docking methodologies, as backbone flexibility adds a huge number of degrees of freedom to the search space, and therefore considerably increases the running time of docking algorithms. Normal mode analysis permits description of protein flexibility as a linear combination of discrete movements (modes). Low-frequency modes usually describe the large-scale conformational changes of the protein. Therefore, many docking methods model backbone flexibility by using only few modes, which have the lowest frequencies. However, studies show that due to molecular interactions, many proteins also undergo local and small-scale conformational changes, which are described by high-frequency normal modes. Here we present a new method, FiberDock, for docking refinement which models backbone flexibility by an unlimited number of normal modes. The method iteratively minimizes the structure of the flexible protein along the most relevant modes. The relevance of a mode is calculated according to the correlation between the chemical forces, applied on each atom, and the translation vector of each atom, according to the normal mode. The results show that the method successfully models backbone movements that occur during molecular interactions and considerably improves the accuracy and the ranking of rigid-docking models of protein-protein complexes. A web server for the FiberDock method is available at: http://bioinfo3d.cs.tau.ac.il/FiberDock.
当蛋白质结合时,它们会发生构象变化。这些变化常常使刚体对接方法无法根据其蛋白质的未结合构象预测复合物的 3D 结构。处理蛋白质骨架灵活性是对接方法的主要挑战,因为骨架灵活性为搜索空间增加了大量自由度,从而大大增加了对接算法的运行时间。正常模式分析允许将蛋白质的灵活性描述为离散运动(模式)的线性组合。低频模式通常描述蛋白质的大规模构象变化。因此,许多对接方法通过仅使用具有最低频率的少数模式来模拟骨架灵活性。然而,研究表明,由于分子相互作用,许多蛋白质也会发生局部和小规模的构象变化,这些变化由高频正常模式来描述。在这里,我们提出了一种新的对接精化方法 FiberDock,该方法通过使用无限数量的正常模式来模拟骨架灵活性。该方法沿着最相关的模式迭代地最小化柔性蛋白质的结构。根据正常模式,根据施加在每个原子上的化学力与每个原子的平移向量之间的相关性,来计算模式的相关性。结果表明,该方法成功地模拟了分子相互作用过程中发生的骨架运动,并大大提高了蛋白质-蛋白质复合物刚性对接模型的准确性和排名。FiberDock 方法的 Web 服务器可在以下网址获得:http://bioinfo3d.cs.tau.ac.il/FiberDock。