Siemens Corporate Research, 755 College Road East, Princeton, NJ, USA.
BMC Bioinformatics. 2010 Jan 15;11:33. doi: 10.1186/1471-2105-11-33.
With the rapid expansion of DNA sequencing databases, it is now feasible to identify relevant information from prior sequencing projects and completed genomes and apply it to de novo sequencing of new organisms. As an example, this paper demonstrates how such extra information can be used to improve de novo assemblies by augmenting the overlapping step. Finding all pairs of overlapping reads is a key task in many genome assemblers, and to this end, highly efficient algorithms have been developed to find alignments in large collections of sequences. It is well known that due to repeated sequences, many aligned pairs of reads nevertheless do not overlap. But no overlapping algorithm to date takes a rigorous approach to separating aligned but non-overlapping read pairs from true overlaps.
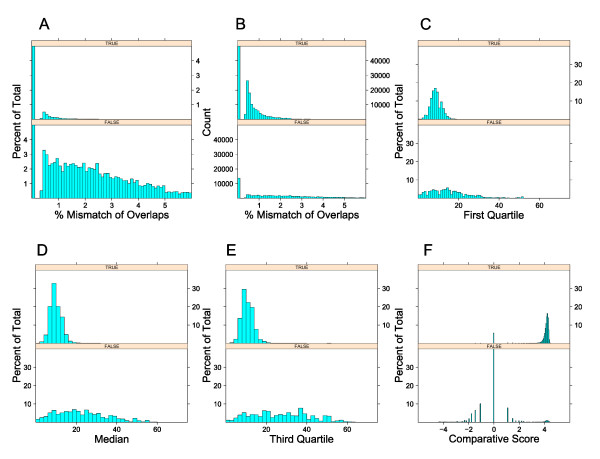
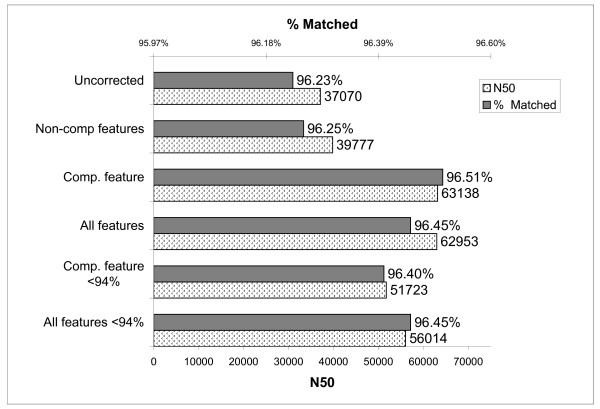
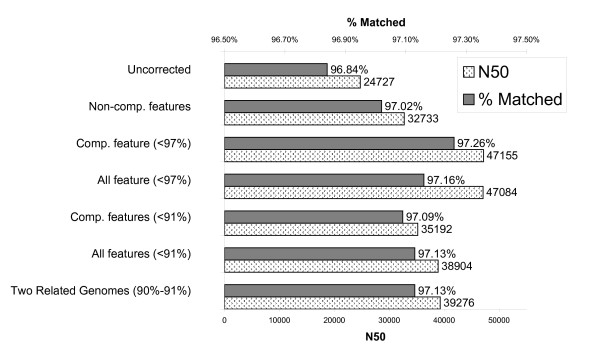
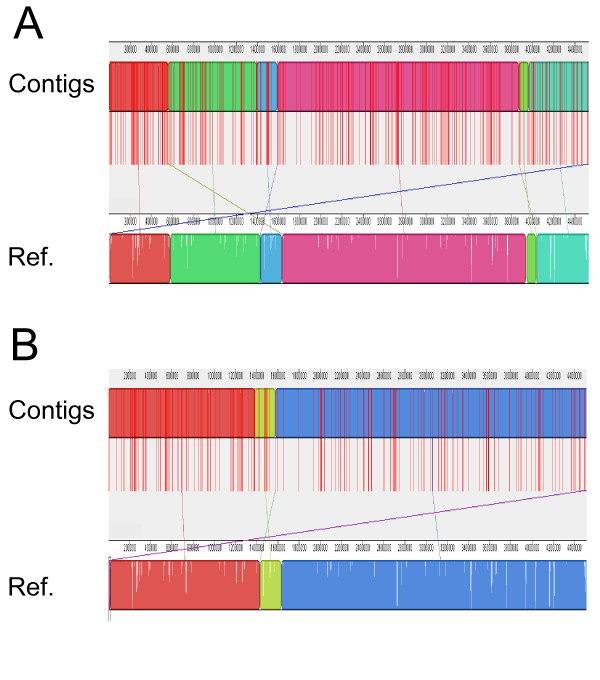
We present an approach that extends the Minimus assembler by a data driven step to classify overlaps as true or false prior to contig construction. We trained several different classification models within the Weka framework using various statistics derived from overlaps of reads available from prior sequencing projects. These statistics included percent mismatch and k-mer frequencies within the overlaps as well as a comparative genomics score derived from mapping reads to multiple reference genomes. We show that in real whole-genome sequencing data from the E. coli and S. aureus genomes, by providing a curated set of overlaps to the contigging phase of the assembler, we nearly doubled the median contig length (N50) without sacrificing coverage of the genome or increasing the number of mis-assemblies.
Machine learning methods that use comparative and non-comparative features to classify overlaps as true or false can be used to improve the quality of a sequence assembly.
随着 DNA 测序数据库的快速扩张,现在可以从先前的测序项目和已完成的基因组中识别相关信息,并将其应用于新生物体的从头测序。本文以实例演示了如何通过增加重叠步骤来利用这些额外信息来改进从头组装。在许多基因组组装器中,找到所有重叠读取对是一项关键任务,为此,已经开发出了高效的算法来在大型序列集合中找到比对。众所周知,由于重复序列的存在,许多对齐的读取对实际上并不重叠。但是,迄今为止,没有任何重叠算法严格区分对齐但不重叠的读取对与真正的重叠。
我们提出了一种方法,通过数据驱动的步骤将 Minimus 组装器扩展,以便在构建连续体之前将重叠分类为真实或虚假。我们在 Weka 框架内使用从先前测序项目中获得的各种读取重叠派生的各种统计信息,在多个分类模型中进行了训练。这些统计信息包括重叠中的错配百分比和 k-mer 频率,以及从将读取映射到多个参考基因组的比较基因组得分。我们表明,在来自大肠杆菌和金黄色葡萄球菌基因组的真实全基因组测序数据中,通过为组装器的连续体阶段提供一组经过整理的重叠,我们将中位数连续体长度(N50)提高了近一倍,而不会牺牲基因组的覆盖率或增加错误组装的数量。
使用比较和非比较特征来将重叠分类为真实或虚假的机器学习方法可以用于提高序列组装的质量。