Ghodsi Mohammadreza, Hill Christopher M, Astrovskaya Irina, Lin Henry, Sommer Dan D, Koren Sergey, Pop Mihai
Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland, USA.
BMC Res Notes. 2013 Aug 22;6:334. doi: 10.1186/1756-0500-6-334.
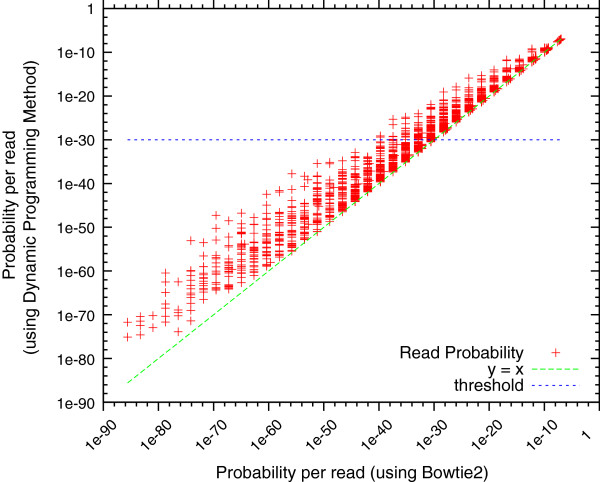
The current revolution in genomics has been made possible by software tools called genome assemblers, which stitch together DNA fragments "read" by sequencing machines into complete or nearly complete genome sequences. Despite decades of research in this field and the development of dozens of genome assemblers, assessing and comparing the quality of assembled genome sequences still relies on the availability of independently determined standards, such as manually curated genome sequences, or independently produced mapping data. These "gold standards" can be expensive to produce and may only cover a small fraction of the genome, which limits their applicability to newly generated genome sequences. Here we introduce a de novo probabilistic measure of assembly quality which allows for an objective comparison of multiple assemblies generated from the same set of reads. We define the quality of a sequence produced by an assembler as the conditional probability of observing the sequenced reads from the assembled sequence. A key property of our metric is that the true genome sequence maximizes the score, unlike other commonly used metrics.
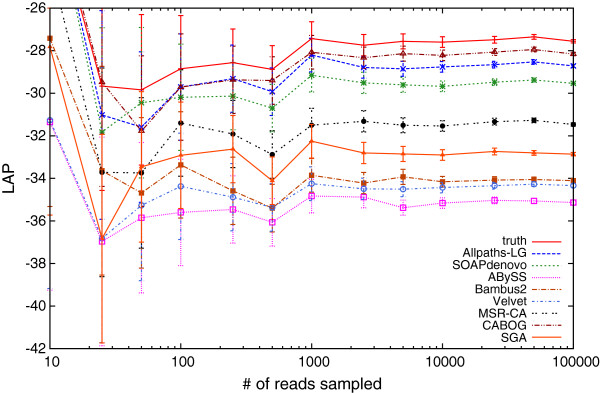
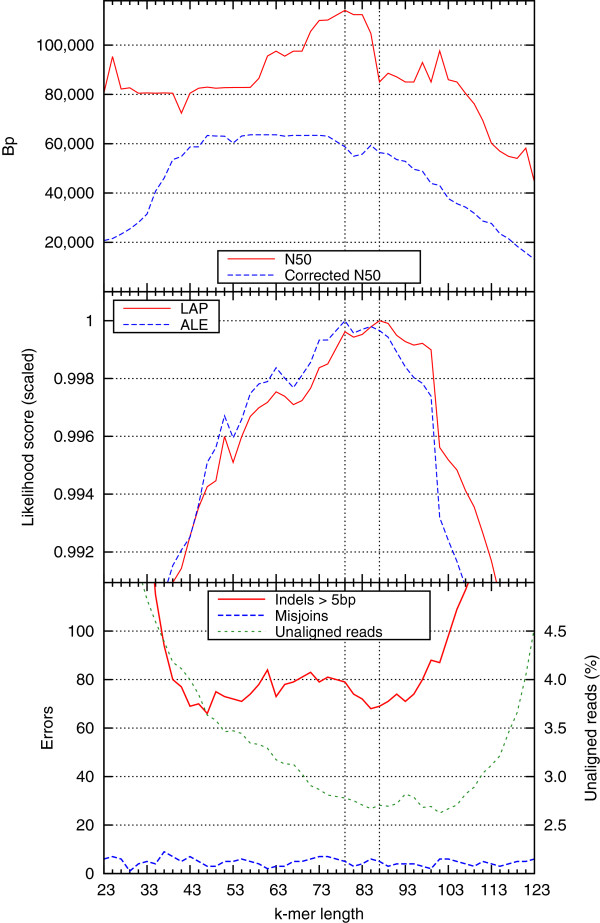
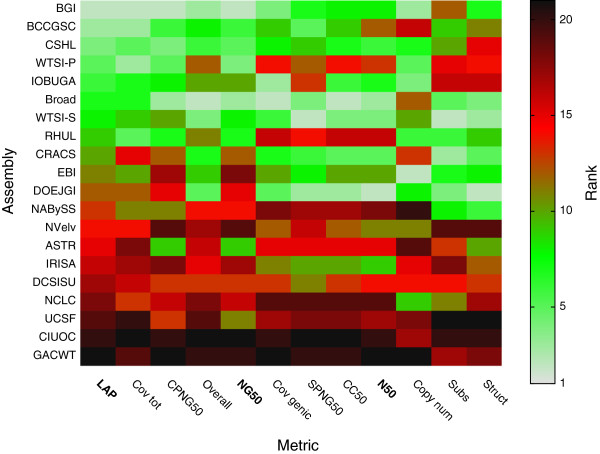
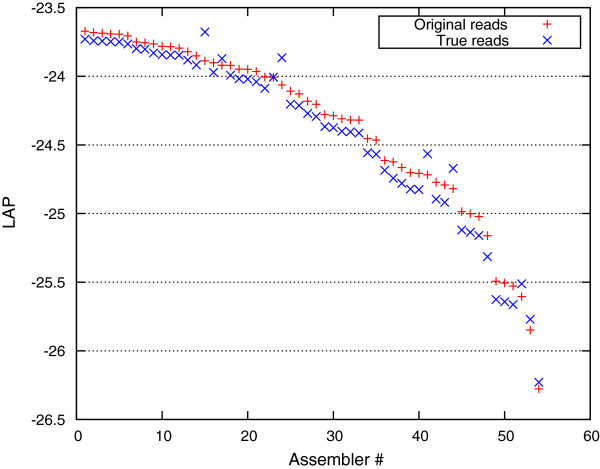
We demonstrate that our de novo score can be computed quickly and accurately in a practical setting even for large datasets, by estimating the score from a relatively small sample of the reads. To demonstrate the benefits of our score, we measure the quality of the assemblies generated in the GAGE and Assemblathon 1 assembly "bake-offs" with our metric. Even without knowledge of the true reference sequence, our de novo metric closely matches the reference-based evaluation metrics used in the studies and outperforms other de novo metrics traditionally used to measure assembly quality (such as N50). Finally, we highlight the application of our score to optimize assembly parameters used in genome assemblers, which enables better assemblies to be produced, even without prior knowledge of the genome being assembled.
Likelihood-based measures, such as ours proposed here, will become the new standard for de novo assembly evaluation.
基因组学领域当前的变革得益于被称为基因组组装软件的工具,这些工具将测序机器“读取”的DNA片段拼接成完整或近乎完整的基因组序列。尽管在该领域进行了数十年的研究并开发了数十种基因组组装软件,但评估和比较组装后的基因组序列质量仍依赖于独立确定的标准的可用性,例如人工编辑的基因组序列或独立生成的映射数据。这些“金标准”的生成成本可能很高,而且可能只覆盖基因组的一小部分,这限制了它们对新生成的基因组序列的适用性。在此,我们引入了一种从头计算组装质量的概率度量方法,该方法允许对从同一组读数生成的多个组装结果进行客观比较。我们将组装软件生成的序列质量定义为从组装序列中观察到测序读数的条件概率。我们的度量方法的一个关键特性是,与其他常用度量方法不同,真实的基因组序列会使分数最大化。
我们证明,即使对于大型数据集,通过从相对较小的读数样本估计分数,我们的从头计算分数也可以在实际环境中快速准确地计算出来。为了证明我们分数的优势,我们用我们的度量方法测量了在GAGE和组装马拉松1组装“竞赛”中生成的组装结果的质量。即使不知道真实的参考序列,我们的从头计算度量方法也与研究中使用的基于参考的评估度量方法紧密匹配,并且优于传统上用于测量组装质量的其他从头计算度量方法(如N50)。最后,我们强调了我们的分数在优化基因组组装软件中使用的组装参数方面的应用,这使得即使在没有待组装基因组的先验知识的情况下也能产生更好的组装结果。
基于似然性的度量方法,如我们在此提出的方法,将成为从头组装评估的新标准。