Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD, USA.
BMC Bioinformatics. 2011 Apr 13;12:95. doi: 10.1186/1471-2105-12-95.
Next-generation sequencing technologies allow genomes to be sequenced more quickly and less expensively than ever before. However, as sequencing technology has improved, the difficulty of de novo genome assembly has increased, due in large part to the shorter reads generated by the new technologies. The use of mated sequences (referred to as mate-pairs) is a standard means of disambiguating assemblies to obtain a more complete picture of the genome without resorting to manual finishing. Here, we examine the effectiveness of mate-pair information in resolving repeated sequences in the DNA (a paramount issue to overcome). While it has been empirically accepted that mate-pairs improve assemblies, and a variety of assemblers use mate-pairs in the context of repeat resolution, the effectiveness of mate-pairs in this context has not been systematically evaluated in previous literature.
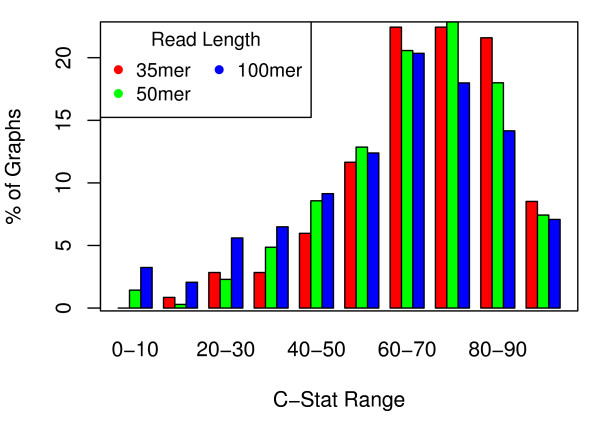
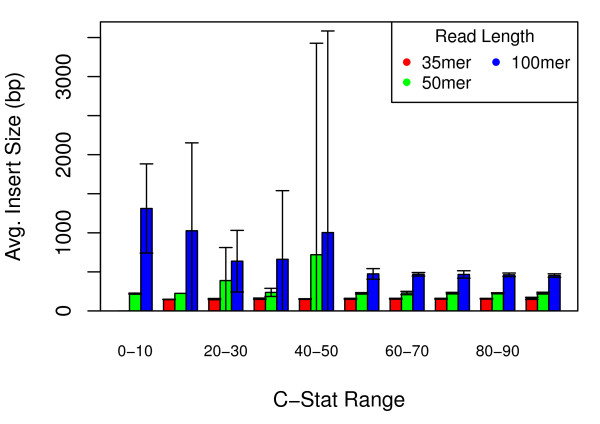
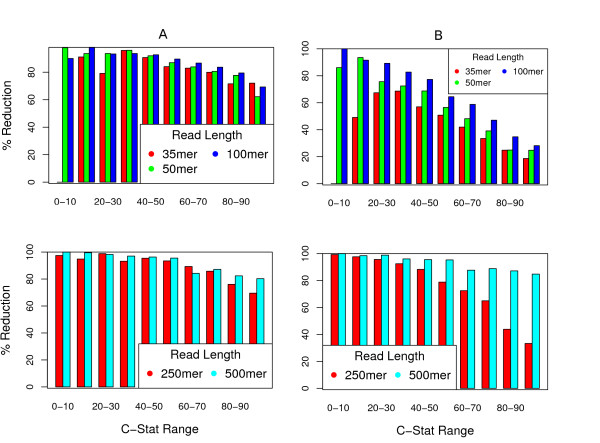
We show that, in high-coverage prokaryotic assemblies, libraries of short mate-pairs (about 4-6 times the read-length) more effectively disambiguate repeat regions than the libraries that are commonly constructed in current genome projects. We also demonstrate that the best assemblies can be obtained by 'tuning' mate-pair libraries to accommodate the specific repeat structure of the genome being assembled - information that can be obtained through an initial assembly using unpaired reads. These results are shown across 360 simulations on 'ideal' prokaryotic data as well as assembly of 8 bacterial genomes using SOAPdenovo. The simulation results provide an upper-bound on the potential value of mate-pairs for resolving repeated sequences in real prokaryotic data sets. The assembly results show that our method of tuning mate-pairs exploits fundamental properties of these genomes, leading to better assemblies even when using an off -the-shelf assembler in the presence of base-call errors.
Our results demonstrate that dramatic improvements in prokaryotic genome assembly quality can be achieved by tuning mate-pair sizes to the actual repeat structure of a genome, suggesting the possible need to change the way sequencing projects are designed. We propose that a two-tiered approach - first generate an assembly of the genome with unpaired reads in order to evaluate the repeat structure of the genome; then generate the mate-pair libraries that provide most information towards the resolution of repeats in the genome being assembled - is not only possible, but likely also more cost-effective as it will significantly reduce downstream manual finishing costs. In future work we intend to address the question of whether this result can be extended to larger eukaryotic genomes, where repeat structure can be quite different.
下一代测序技术使得基因组的测序比以往任何时候都更快、更便宜。然而,随着测序技术的改进,从头组装基因组的难度也增加了,这在很大程度上是由于新技术产生的较短读长。使用配对序列(称为 mate-pairs)是一种标准的方法,可以通过不依赖于手动完成来区分组装,从而获得更完整的基因组图谱。在这里,我们研究了 mate-pairs 信息在解决 DNA 中的重复序列(一个需要克服的主要问题)方面的有效性。虽然已经从经验上接受了 mate-pairs 可以改善组装,并且各种组装器在重复分辨率的上下文中使用 mate-pairs,但在以前的文献中,没有系统地评估 mate-pairs 在这种情况下的有效性。
我们表明,在高覆盖率的原核组装中,短 mate-pairs 文库(约为读长的 4-6 倍)比当前基因组项目中构建的文库更有效地解决重复区域的歧义。我们还证明,通过“调整”mate-pairs 文库以适应正在组装的基因组的特定重复结构,可以获得最佳的组装-可以通过使用未配对的读取进行初始组装来获得该信息。这些结果是在对 360 个“理想”原核数据的模拟以及使用 SOAPdenovo 对 8 个细菌基因组进行组装的基础上得到的。模拟结果提供了 mate-pairs 在解决真实原核数据集重复序列方面的潜在价值的上限。组装结果表明,我们调整 mate-pairs 的方法利用了这些基因组的基本特性,即使在存在碱基调用错误的情况下,使用现成的组装器也可以获得更好的组装。
我们的结果表明,通过将 mate-pairs 的大小调整到基因组的实际重复结构,可以显著提高原核基因组组装的质量,这表明可能需要改变测序项目的设计方式。我们提出,一种两级方法-首先使用未配对的读取生成基因组的组装,以评估基因组的重复结构;然后生成提供有关组装基因组中重复分辨率的最信息的 mate-pairs 文库-不仅是可能的,而且可能更具成本效益,因为它将大大降低下游手动完成的成本。在未来的工作中,我们打算解决这个问题,即这个结果是否可以扩展到重复结构可能大不相同的更大的真核基因组。